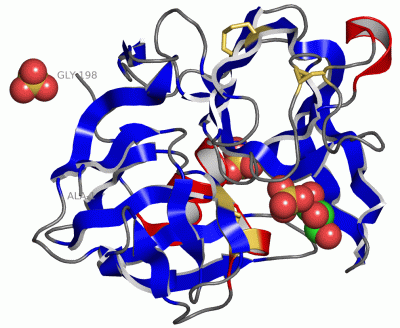
|
|
|
|
 Description
Description|
|
Compounds
|
||||||||||||||||||||||||||||||||||||||||||||
Chains, Units
Summary Information (see also Sequences/Alignments below) |
Ligands, Modified Residues, Ions (2, 4)| Asymmetric/Biological Unit (2, 4) |
Sites (4, 4)
Asymmetric Unit (4, 4)
|
SS Bonds (3, 3)
Asymmetric/Biological Unit
|
||||||||||||||||
Cis Peptide Bonds (1, 1)
Asymmetric/Biological Unit
|
||||||||
SAPs(SNPs)/Variants (0, 0)| (no "SAP(SNP)/Variant" information available for 1QRX) |
PROSITE Motifs (2, 2)
Asymmetric/Biological Unit (2, 2)
|
||||||||||||||||||||||||||||||||
Exons (0, 0)| (no "Exon" information available for 1QRX) |
Sequences/Alignments
Asymmetric/Biological UnitChain A from PDB Type:PROTEIN Length:198 aligned with PRLA_LYSEN | P00778 from UniProtKB/Swiss-Prot Length:397 Alignment length:198 209 219 229 239 249 259 269 279 289 299 309 319 329 339 349 359 369 379 389 PRLA_LYSEN 200 ANIVGGIEYSINNASLCSVGFSVTRGATKGFVTAGHCGTVNATARIGGAVVGTFAARVFPGNDRAWVSLTSAQTLLPRVANGSSFVTVRGSTEAAVGAAVCRSGRTTGYQCGTITAKNVTANYAEGAVRGLTQGNACMGRGDSGGSWITSAGQAQGVMSGGNVQSNGNNCGIPASQRSSLFERLQPILSQYGLSLVTG 397 SCOP domains d1qrxa_ A: alpha-Lytic protease SCOP domains CATH domains --1qrxA01 A:3-76,A:184-196 Trypsin-like serine proteases 1qrxA02 A:77-183 Trypsin-like serine proteases 1qrxA01 -- CATH domains Pfam domains ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------ Pfam domains SAPs(SNPs) ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------ SAPs(SNPs) PROSITE -------------------------------TRYPSI---------------------------------------------------------------------------------------------------TRYPSIN_SER -------------------------------------------------- PROSITE Transcript ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------ Transcript 1qrx A 1 ANIVGGIEYSINNASLCSVGFSVTRGATKGFVTAGHCGTVNATARIGGAVVGTFAARVFPGNDRAWVSLTSAQTLLPRVANGSSFVTVRGSTEAAVGAAVCRSGRTTGYQCGTITAKNVTANYAEGAVRGLTQGNACMGRGDSGGSWITSAGQAQGVMSGGNVQSNGNNCGIPASQRSSLFERLQPILSQYGLSLVTG 198 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190
|
||||||||||||||||||||
SCOP Domains (1, 1)
Asymmetric/Biological Unit
 |
CATH Domains (1, 2)
Asymmetric/Biological Unit
|
Pfam Domains (0, 0)| (no "Pfam Domain" information available for 1QRX) |
Gene Ontology (6, 6)|
Asymmetric/Biological Unit(hide GO term definitions) Chain A (PRLA_LYSEN | P00778)
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
Interactive Views
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Still Images
|
||||||||||||||||
Databases
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Analysis Tools
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Entries Sharing at Least One Protein Chain (UniProt ID)
Related Entries Specified in the PDB File
|
|