![]()
Comprehensive Bending Classification of Nucleic Acid Double Helix Structures
(Last update: November 15, 2004)
Peter Slickers, Kristina Mehliß, Jan Reichert, Jürgen Sühnel
Biocomputing Group, Leibniz Institute for Age Research -
Fritz Lipmann Institute (formerly known as Institute of Molecular Biotechnology),
Jena / Germany
Introduction
The aim of this classification is to provide quantitative geometrical information on bending properties of nucleic acid double helix structures adopting a uniform analysis approach. The notion bending is used to indicate any global deviation from a straight helix axis. An abrupt local alteration of the helix axis direction is called a kink. The helix axis can, however, also show a smooth bending. Bending can be either an intrinsic property or may be enforced by outer factors like proteins or other ligands, crystal packing, asymmetric charge neutralization of backbone phosphate groups, cation/anion localization or crosslinking. Bending is in any case reflected in local base or dinucleotide step parameters, like roll and tilt angles.
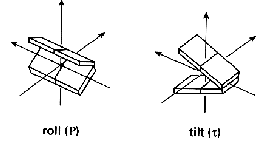
However, this local analysis is not sufficient. Local variations may cancel each other and result in a straight helix axis. Therefore, a thorough bending analysis of nucleic acid double helix structures has to include both the local and global point of view.
More information on geometrical nucleic acid helix
parameters can be found here.
Method
The Double
Helix/ Bending Analysis web tool included in the IMB Jena Image Library of
Biological Macromolecules offers information on local and global
helical parameters. The parameters provided include selected
information on base step geometries and on backbone conformations and
groove widths. In addition, full outputs from the programs CURVES [1]
and FREEHELIX [2] are
available. For the global bending analysis the helical axis is first
determined with the CURVES algorithm
[1]. The nucleic acid structures are then oriented in such a way
that the coordinate axes are parallel to the principal axes of inertia
of the curvilinear helical axis. This results in an immediate visual
impression on helix axis bending. Subsequently, the following
geometrical models are fitted to the helical axis.
model |
geometry parameters |
minimum number of base pairs |
independent number of fit parameters |
plane |
- |
- |
4 |
straight line |
- |
6 |
3 |
| circular line (arc) | radius of curvature: r | 7 |
6 |
| single-kink line | kink angle: a; length of segments: l1, l2 |
8 |
7 |
| double-kink line | kink angles: a1, a2;
twist angle: o; lengths of segments : l1, l2, l3 |
13 |
10 |
The definition of the geometrical parameters is described here. Fitting is done with the program ARC_FIT written by P. Slickers. The most appropriate models are selected according to the goodness of fit sigma2.
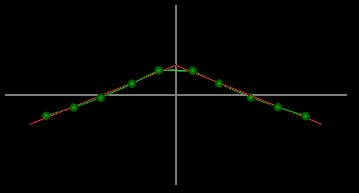
It is important to note that a structure with a helical
axis of the type shown in the image is classified as a single-kink
duplex.
The Data Set
The analysis is currently done for more than 1201
nucleic acid double helix structures with at least six base pairs. They
include free nucleic acids, nucleic acid-protein complexes and
complexes of nucleic acids with small ligands. The global shape of the
helical axis is a rather crude geometrical measure. We have therefore
included high and low-resolution diffraction structures and, in
addition, model and NMR structures. Given for NMR structures more than
one model is provided only the first model is included in the analysis.
Z-DNA structures (Nucleic Acid Database code: z.....), unusual nucleic
acids (Nucleic Acid Database code: u......) and further structures
with one or more nucleotides in syn conformation are not
taken into account. A few additional unusual structures have been
removed manually. A complete listing and structure compilations listed
by base pair number and protein type can be found here. The database us updated on a regular basis.
Limitations
Bending Classification of Nucleic Acid Double Helix Structures
The following classification is done according to the
geometrical shape of the helical axis. Structures with a
goodness-of-fit value > 2 are categorized as other. Note that this classification is inevitably
arbitrary to some extent.
a) Among these structures there are 159 duplexes with 6 base pairs. In this case no other model than a straight line can be fitted.
Contrary to the bending classification given above the
following classification is done taking into account the extent of
bending alone independent of the appropriate models describing the
helical axis. The following generalized measure of bending M has been
used:
model |
M corresponds to |
comment |
| circular line | kink angle: a (radius of curvature: r) |
From circular line structures for which a single-kink line has an almost equal goodness of fit a quantitative relationship between the radius of curvature and the kink angle has been derived:
|
| single-kink line | kink angle: a | |
| double-kink line | classification I - sum of kink angles: a1 + a2; classification II - the largest kink angle: either a1 or a2, max(a1,a2) |
This classification is done independent of the value of the twist angle o. |
The bending class no bending is identical to the
straight-line case described above.
| extent of bending | classification | bending measure M | number of structures |
| no bending | classification I=II (HTML | TXT) | M = 0 ° |
classification I: 322 classification II: 322 |
| slight bending | classification I (HTML
| TXT)
classification II (HTML | TXT) |
0 ° < M <= 10 ° |
classification I: 208 classification II: 226 |
| moderate bending | classification I (HTML
| TXT)
classification II (HTML | TXT) |
10 ° < M <= 45 ° |
classification I: 535 classification II: 580 |
| strong bending | classification I (HTML
| TXT)
classification II (HTML | TXT) |
M > 45 ° |
classification I: 136 classification II: 73 |
total 1201 |
|||
| The complete classification in one text
file including nucleic acid and protein descriptions (last update on November 15, 2004).
(Use the grep command for generating subsets). |
classification I
(sum of kink angles for the double-kink model)
version A
version B
classification II
(largest kink angle for the double-kink model)
version A
version B
|
total 1201 |
References
JenaLib
Home| Helix/Bending Analysis Home| Nucleic Acid Bending
Classification Home
Biocomputing Group, Leibniz Institute for Age Research -
Fritz Lipmann Institute (formerly known as Institute of Molecular
Biotechnology), Jena / Germany
All tools for analyzing individual structures were
developed by Peter Slickers.
The bending classification was done by Kristina
Mehliß and Jürgen
Sühnel with support from Jan Reichert and Friedrich Haubensak.