|
|
 General Aspects
General Aspects
The cis/trans isomerisation of a
peptidyl-prolyl bond leads to a different propagation direction
of the polypeptide backbone in each isomer.
"The cis/trans isomerisation at the peptide bond
N-terminal to proline resembles a molecular
switch with the following characteristics:
1. There are two switch positions, other positions are unstable
2. The energy needed to operate the switch is high on the energy scale
for bio-
logical recognition processes.
3. Operation of the switch leads to an amplification of an effect at
the place
where the signal arrives. In the case of the proline switch this
amplification
manifests itself chiefly in the form of a mechanical movement, which
may
be the movement of a segment of the protein backbone.
4. Isomerisatin catalysts such as the PPIases act as variable
components for
reducing the switch resistant, and in the presents of many switches
they
assure extra selectivity. The switching resistance (rotational
barriers) is
controlled by varying the enzyme concentration and the selection of
switching
elements by means of the enzyme specificy."
[G. Fischer, Angew.
Chem. Int. Ed. Engl. 33(1994)1415]
Peptidyl-prolyl Bonds in Peptides and Proteins
The following data on the occurence of cis prolyl bonds in
proteins were derived from a set
of non-redundant protein structures from the Brookhaven PDB. For
analysing the structural
parameters (peptide bond angle omega, secondary structure) the program
package Iditis
(Oxford Molecular,
Version 2.1, data base 9.0) was used.
The distribution of the peptide bond angle omega
for peptidyl-prolyl bonds in proteins
shows significant peaks at 180 deg. (trans peptide bond) and 0
deg. (cis peptide bond).
There are some differences between the data on the cis
content in Xaa-Pro bonds (Xaa:
amino acid N-terminal to proline) derived from different data bases [Stewart
et al. J.Mol.
Biol.
214(1990)253, McArthur
& Thornton J.Mol.Biol. 218(1991)397, Iditis 6.0, Iditis 9.0,
data from the actual Brookhaven PDB will be added as soon as
possible]. McArthur and
Thornton found three types of peptidyl-prolyl bonds which do not occur
in cis conformation
(Cys, Met, Trp). Stewart et al. showed only Trp-Pro to take no cis
conformation. From the
Iditis 2.1, data base 9.0 data for all amino acids were received. In
some cases the cis content
decreases from 1990 to 1995 (Arg, Leu), in the most cases deviations
show no regularity.
These results teach a careful approach to statistical data derived
from the Brookhaven
PDB.
Investigations on 'peptidyl-prolyl bonds and
secondary structure' show, that trans petidyl-
prolyl bonds are distributed in all types of secondary structure. Cis
peptidyl are found pri-
marily in bends and turns, suggesting a specific structural role for
this type of bonding.
[Stewart
et al. J.Mol.Biol. 214(1990)253].
From NMR investigations on proline containig peptides results a
correlation between
the cis content of
peptidyl-prolyl bonds in peptides and proteins [Reimer
et al. J.Mol.Biol.
279(1998)449].
Peptidyl-prolyl Cis/Trans Isomerases (PPIases)
DefinitionCyclophilins (Cyp) - inhibited by Cyclosporin A (CsA)
FK506 Binding Proteins (FKBP's) - inhibited by FK506
Parvulins
Cyclophilins in the Brookhaven PDB| 1ak4 | HUMAN CYCLOPHILIN A BOUND TO THE N-TERMINAL DOMAIN OF HIV-1 CAPSID PROTEIN |
| 1clh | CYCLOPHILIN (NMR, 12 STRUCTURES) |
| 1cyn | CYCLOPHILIN B COMPLEXED WITH [D-(CHOLINYLESTER)SER8]-CYCLOSPORIN |
| 1fgl | CYCLOPHILIN A COMPLEXED WITH A FRAGMENT OF HIV-1 GAG PROTEIN |
| 1lop | CYCLOPHILIN A COMPLEXED WITH SUCCINYL-ALA-PRO-ALA-P-NITROANILIDE |
| 1mik | CYCLOPHILIN A |
| 1oca | HUMAN CYCLOPHILIN A, UNLIGATED, NMR, 20 STRUCTURES |
| 1rmh | RECOMBINANT CYCLOPHILIN A FROM HUMAN T CELL |
| 2cpl | CYCLOPHILIN A |
| 2cyh | CYCLOPHILIN A COMPLEXED WITH DIPEPTIDE ALA-PRO |
| 2rma | CYCLOPHILIN A (E.C.5.2.1.8) COMPLEXED WITH CYCLOSPORIN A |
| 2rmb | CYCLOPHILIN A (E.C.5.2.1.8) COMPLEXED WITH DIMETHYL-CYCLOSPORIN A |
| 2rmc | CYCLOPHILIN C COMPLEXED WITH CYCLOSPORIN A |
| 3cyh | CYCLOPHILIN A COMPLEXED WITH DIPEPTIDE SER-PRO |
| 3cys | CYCLOPHILIN A COMPLEXED WITH CYCLOSPORIN A (NMR, 22 STRUCTURES) |
| 4cyh | CYCLOPHILIN A COMPLEXED WITH DIPEPTIDE HIS-PRO |
| 5cyh | CYCLOPHILIN A COMPLEXED WITH DIPEPTIDE GLY-PRO |
FKBP's in the Brookhaven PDB| 1bkf | FK506 BINDING PROTEIN FKBP MUTANT R42K/H87V COMPLEX WITHFK506 |
| 1fap | THE STRUCTURE OF THE IMMUNOPHILIN-IMMUNOSUPPRESSANT FKBP12-RAPAMYCINE |
| 1fkb | FK506 BINDING PROTEIN (FKBP) COMPLEX WITH IMMUNOSUPPRESSANT RAPAMYCINE |
| 1fkf | FK506 BINDING PROTEIN (FKBP) COMPLEX WITH IMMUNOSUPPRESSANT FK506 |
| 1fkg | FK506 BINDING PROTEIN (FKBP) COMPLEX WITH ROTAMASE INHIBITOR |
| 1fkh | FK506 BINDING PROTEIN (FKBP) COMPLEX WITH ROTAMASE INHIBITOR |
| 1fki | FK506 BINDING PROTEIN (FKBP) COMPLEX WITH ROTAMASE INHIBITOR |
| 1fkj | ATOMIC STRUCTURE OF FKBP12-FK506 |
| 1fkk | ATOMIC STRUCTURE OF FKBP12, AN IMMUNOPHILIN BINDING PROTEIN |
| 1fkl | ATOMIC STRUCTURE OF FKBP12-RAPAYMYCIN |
| 1fkr | FK506 AND RAPAMYCIN-BINDING PROTEIN (FKBP12) (NMR, 20 STRUCTURES) |
| 1fks | FK506 AND RAPAMYCIN-BINDING PROTEIN (FKBP12) (NMR) |
| 1fkt | FK506 AND RAPAMYCIN-BINDING PROTEIN (FKBP12) (NMR) |
| 1nsg | THE STRUCTURE OF THE IMMUNOPHILIN-IMMUNOSUPPRESSANT FKBP12- RAPAMYCINE |
| 1pbk | HOMOLOGOUS DOMAIN OF HUMAN FKBP25 |
| 1rot | STRUCTURE OF FKBP59-I, THE N-TERMINAL DOMAIN OF A 59 KDA FK506-BINDING PROTEIN |
| 1rou | STRUCTURE OF FKBP59-I, THE N-TERMINAL DOMAIN OF A 59 KDA FK506-BINDING PROYEIN |
| 1tco | TERNARY COMPLEX OF A CALCINEURIN A FRAGMENT, CALCINEURIN B, FKBP12 |
Mechanistical Aspects of the Cis/Trans Isomerisation
Experimental studies of intramolecular catalysis of amide isomerisation
in model systems
shows an evidence for a hydrogen bond between the side chain and the
prolyl imide
nitrogen in a cis peptidomimetic [Cox et al. J.Am.Chem.Soc.
119(1997)2307].
MO and force field calculations on proline containing dipeptides
shows, that the C-ter-
minal amide proton interacts favorably with the imide nitrogen of the
proline moiety.
This calculations indicate a cis/trans barrier lowering of 1.4
kcal/mol due to intramole-
cular catalysis [Fischer & Karplus J.Am.Chem.Soc. 116(1994)11931].
Folding experiments and mutagenic analysis of dihydrofolate reductase
show that the rate
limiting step of refolding, the isomerisation of the proline 66
residue can be intramole-
cularly ctalyzed by the side chain of the arginine 44 residue. The
guanidinium group NH2
nitrogen of this residue forms a hydrogen bond to the imide nitrogen
of the proline
residue.
Metal ions (Lewis Acids) in small amounts can catalyse the the
isomerisation of amides.
The side chain of substituted prolines acts as a binding site for
Cu(II) ions to catalyse the
prolyl isomerisation [Cox et al. J.Am.Chem.Soc. 118(1996)5332].
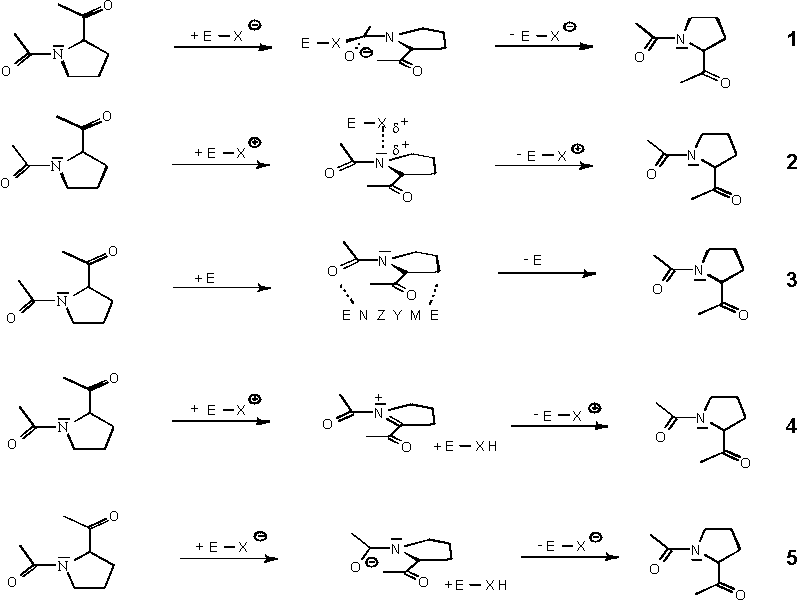
The data discussed above indicate a general acid catalysis of the cis/trans
isomerisation
of a peptidyl-prolyl bond (equation 2 in the scheme).
In the structure of the PPIase cyclophilin compexed with a substrate,
the guanidinium
group of an arginine in the active site can form a hydrogen bond with
the lone pair
electrons of the peptidyl-prolyl bond in the substrate peptide. The
structure of different
complexes of cyclophilin with inihibitors, substrate peptides and
protein fragments may
provide insight into the mechanism of the enzymatic catalysis of the
prolyl isomerisa-
tion.
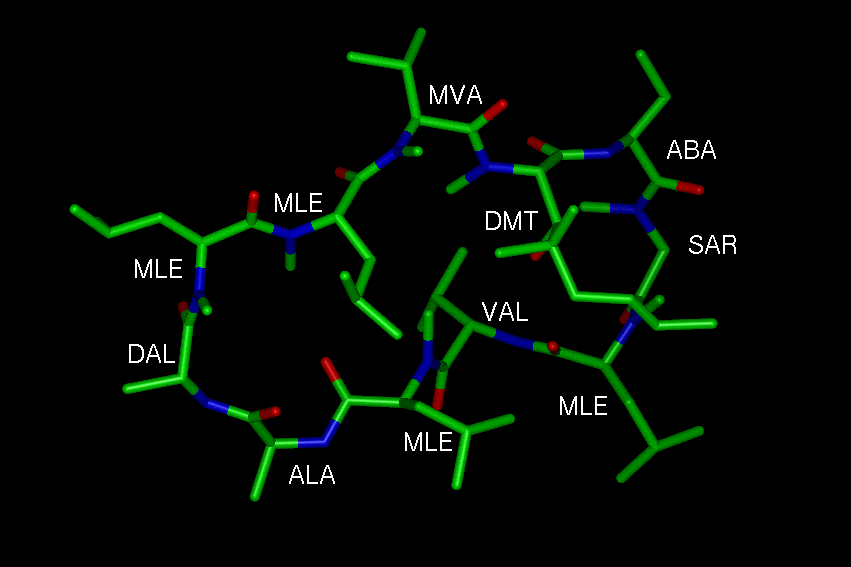
1. The cyclophilin-substrate complex:
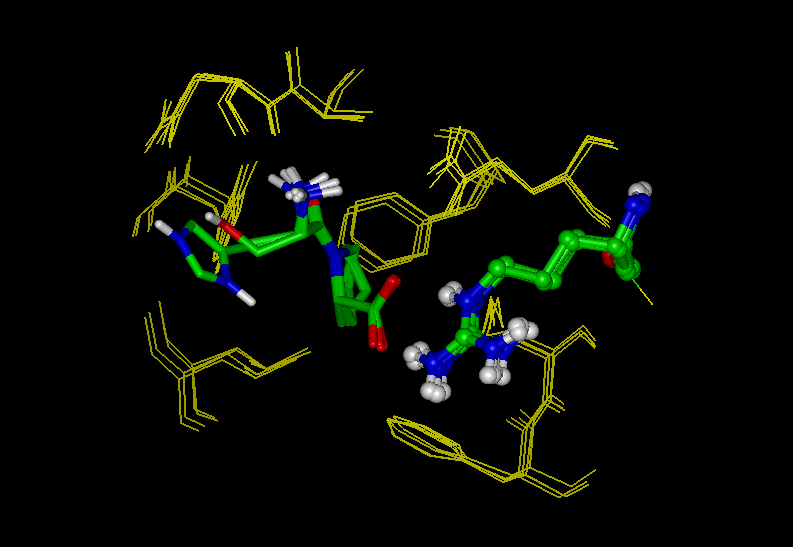
The first three figures show the recombinant cyclophilin A from human T
cell (Cyp)
complexed with the model peptide Suc-Ala-Ala-Pro-Phe-pNA (AAPF);
PDB code: 1rmh
Figure 1: The Cyp-AAPF complex
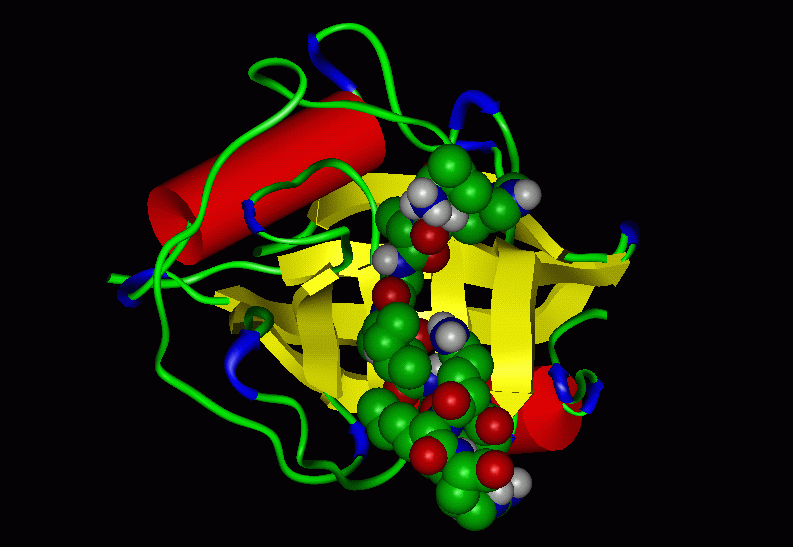
Figure 2: The active site (proline binding
pocket, Conolly surface)
of the Cyp-AAPF complex
Figure 3: The active site (proline binding
pocket, Conolly surface transparent)
of the Cyp-AAPF complex
The following figures shows the recombinant cyclophilin A from human T
cell (Cyp)
complexed with a fragment of the HIV-1 GAG
protein (PDB code 1fgl).
This protein
play an essential role in the replication of the
HIV (movie from the Microbiology Video
Library at the Department of
Microbiology & Immunology, University of Leicester).
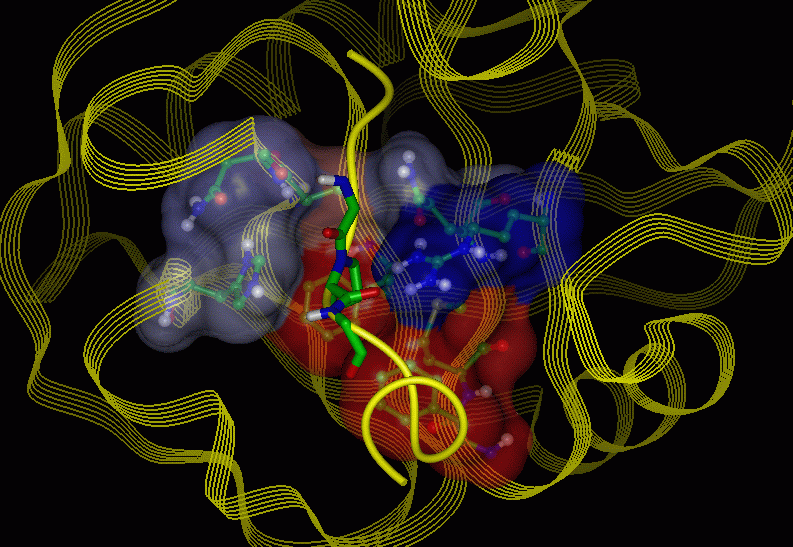
Figure 4: The HIV-1 GAG-fragment - Cyp complex
Figure 5: The active site (proline binding
pocket, Conolly surface, the fragment
of the HIV-1 GAG protein is shown as a yellow colored solid ribbon)
of this complex
Figure 6: The active site (proline binding
pocket, Conolly surface transparent)
of this complex
The side chain of the substrate proline sits in the hydrophobic pocket
made up of the side
chains of Phe60, Met61, Phe113, Ile122 (red colored region on the
Conolly surface). The
Arg55 residue hydrogen bonds to the lone pair electrons of the amide
nitrogen. Zhao & Ke
[Biochemistry
35(1996)7356] proposed on the basis of the crystal structure, that
the hydro-
gen bond deconjugates the resonance of the amide bond during
catalysis. The C-terminal
region of the proline interacts with hydrophilic amino acids (Arg55,
His126, blue colored
region on the Conolly surface). These facts provide the mechanism
shown in equation 2 in
the scheme discussed mentioned above.
The mutant cyclophilin protein in which the arginine residue is
replaced by alanine (R55A)
shows dramatically lower PPIase activity below 1% of the wild type
enzyme [Zydowski
et al. Protein Science 1(1992)1092]. If the histidine residue in the
active site is replaced
by glutamine (H126Q), the PPIase activity decreases (0.5% of wild type
activity) [Zydows-
ki et al.].
A general acid catalysis of the enzymatic cis/trans
isomerisation (for cyclophilin) by Arg55
is not in conflict with the observed pH independence [Harrison
& Stein Biochemistry 29
(1990)3813].
The Arg55 is expected to be protonated at a pH range of 5.5-9.0.
2. The position of Arg55 in different complexes
[derived from Zhao & Ke ; Biochemistry
35(1996)7356]
The structures of the cyclophilin complexed with the dipeptides
Ala-Pro, Ser-Pro, His-Pro
and Gly-Pro (2cyh, 3cyh, 4cyh, 5cyh)
are very similar to the unligated protein (2cpl).
The
superposition of the amino acids forming the proline binding site from
uncomplexed cyclo-
philin (2cpl)
and the protein ligated with the Ala-Pro (2cyh)
dipeptide revealed only small
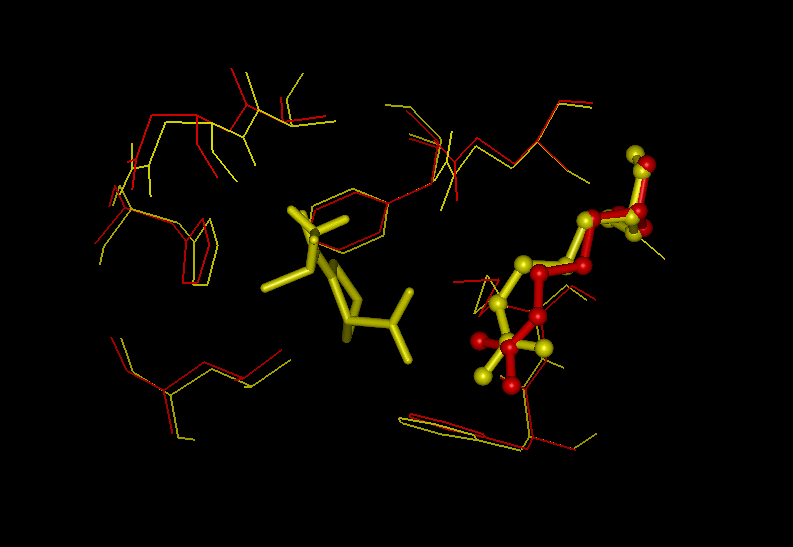
displacements (Fig. 7)
Figure 7: Superposition of the active sites in
Cyp and the complex Cyp-AlaPro
(2cpl: red, 2cyh: yellow)
In all complexes the two carboxy-terminal oxygens of the dipeptide form
hydrogen bonds to
the Arg55 (Fig. 8). The position of the Arg55 in the cyclophilin
complexed with cyclosporin A (2rma)
is very similar to the structures discussed above (Fig. 9).
Figure 8: Superposition of all dipeptide-Cyp
structures
(dipeptides [stick] and Arg55 [ball & stick] colored by atoms)
Figure 9: Superposition of the Cyp-AlaPro (yellow) and the Cyp-cyclosporin A complex (red)
The overall binding of the dipeptides to cyclophilin also closely
resembles that of the substrate
tetrapeptide Suc-AlaAlaProPhe-pNA (AAPF) and the binding region of the
HIV-1 GAG pro-
tein. In these cases, the residue Arg55 form one hydrogen bond to the
carbonyl oxygen an one to
the imide nitrogen atom of the substrate proline (Fig. 10).
Figure 10: Superposition of the two
Cyp-substrate structures
(Cyp-AAPF yellow, Cyp-HIV-1 GAG protein red)
In comparision to the dipeptide binding the hydrogen bonding pattern
of the proline, the orienta-
tion of the proline in the proline binding pocket and the conformation
of the side chain of Arg55
is changed (Fig.11).
Figure 11: Superposition of the Cyp-AAPF (red)
and the Cyp-AlaPro
(yellow, proline is colored by atoms)
Only in the cyclophilin-substrate structures, the guanidinium group of
Arg55 interacts via hydro-
gen bond with the imide nitrogen of the substrate proline.
The similarity between the orientation of the active side residues in
cyclophilin, the cyclophilin-
cyclosporin A complex (inhibitor) and the dipeptide complexes imply
these questions:
"Are dipeptides inhibitors for cyclophilins or do they have a
different catalytic mechanism from
the substrate-petides AAPF and HIV-1 GAG fragment? [Zhao &
Ke]"
Which role play the amino acid Arg55 in the catalytic mechanism of this
enzyme?
Which role play the amino acid His126 in the catalytic mechanism of
this enzyme?
|
|
|
|
Beutenbergstraße 11 D-07745 Jena • Germany |
Phone: +49 3641 65-6000 Fax: +49 3641 65-6351 |
E-mail: info@leibniz-fli.de www.leibniz-fli.de |
Data Privacy Imprint |
 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}