|
|
 General Aspects
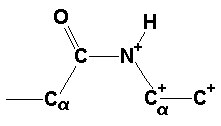
General AspectsIn protein structures, the peptide bond conformation is found to be trans in the absolute majority of the cases (Ramachandran & Sasisekharan, 1968). An exception to this constitute the peptide bonds between any amino acid and Pro (Xaa-Pro), for which an appreciable fraction occurs in the cis conformation. A survey by Stewart et al. (1990) found only 0.05% of all Xaa-nonPro, but 6.5% of all Xaa-Pro peptide bonds to occur in the cis conformation, MacArthur and Thornton (1991) found 5.5% cis for Xaa-Pro and recently we (Weiss et al., 1998a) found in a much larger non-redundant set of 571 proteins 5.2% cis for Xaa-Pro and 0.03% cis for Xaa-nonPro.
The reason for this preponderance of the trans peptide bond is believed to lie in the energy difference between the two different forms. It is clear that the cis form is energetically less stable due to a steric repulsion of the two neighbouring Calpha atoms, but the absolute numbers have been the basis of much debate over the years. Experimentally, Radzicka et al. (1988) found that the model compound N-methylacetamide occurs at about 1.5% in the cis form, regardless of the solvent. From this, a difference in free enthalpy of 2.4 kcal/mol can be computed. Almost the same value was reported by Drakenberg and Forsén [J.Cem.Soc.,Chem.Comm. 1971, 1404-1405]. They found deltaG = 2.5 (+/- 0.4 kcal/mol in water. For Pro-containing peptides, Grathwohl and Wüthrich (1976) reported a cis content of about 10 - 15% when the charge at the C-terminus is removed either by protonation or by a protecting group. This corresponds to a free enthalpy difference between cis and trans of about 1.0 - 1.3 kcal/mol at 293 K.
Theoretical calculations are pretty much in accord with these experimental values. Maigret et al. (1970) reported a 0.5 kcal/mol difference between cis and trans for Acetyl-Pro-N(CH3) based on quantum mechanical calculations, and Jorgensen and Gao [J.Am.Chem.Soc. 1988, 110, 4212-4216] reported 2.5 kcal/mol difference for the model compound N-methylacetamide in the gas phase and 2.6 kcal/mol in water. Since these numbers depend critically on the geometrical and force field parameters used, they have to be taken with care.
In proteins, the energetic situtation is more difficult to describe and therefore less clear. Ramachandran and Mitra (1976) used conformational energy calculations of tripeptide units to derive expected frequencies of 0.1% and 30% cis for an Ala-Ala and an Ala-Pro peptide bond respectively. These numbers correspond to respective enthalpy differences of 4.0 kcal/mol and 0.5 kcal/mol.
Experimental data for proteins stem mainly from cis-Pro point mutations. Tweedy et al. (1993) found that the Pro202->Ala mutant of carbonic anhydrase is by about 5 kcal/mol less stable than the wild type enzyme. In both the wild type and the mutant enzyme the peptide bond between residues 201 and 202 oocurs in the cis conformation; therefore the decrease in stability of the protein upon mutating Pro202 to Ala is thought to be mainly due to the less favorable cis/trans equilibrium, although the authors say that other factors may also contribute. Schultz and Baldwin (1992) reported a destabilization of 2.7 kcal/mol for the Pro93->Ala mutant of ribonuclease A with respect to the wild type protein. In the three-dimensional structure of this mutant (Pearson et al., 1998) the loop containing the Tyr92-Ala93 cis peptide bond becomes more mobile, but it seems as if the cis conformation is retained. Mayr et al. (1993) also found a very strong destabilization of about 5 kcal/mol for the Pro39->Ala mutant of ribonuclease T1, but in this case it's not clear whether the mutated protein still contains the peptide bond in the cis conformation.
Due to the double bond character of the amide bond, an appreciable barrier exists for the rotation around the C-N bond. Experimental results for the activation enthalpy for model compounds (Drakenberg & Forsén, 1971) and theoretical calculations ([Christensen et al., J.Chem.Phys. 1970, 53, 3912-3922]; [Perricaudet & Pullman, 1973]) agree on values of about 20 kcal/mol for the activation enthalpy. Such a large barrier makes the interconversion between the two conformations a rather slow process at room temperature. If one assumes for simple stereochemical reasons that all peptide bonds are synthesized in the same conformation, which must be obviously the trans conformation, then it becomes clear that isomerization must occur at some stage of the folding process of the protein, if the correctly folded protein contains at least one peptide bond in cis conformation. And indeed it has been demonstrated (Brandts et al., 1975) that isomerization of the peptide bonds before proline residues plays a decisive role in protein folding. The discovery of prolyl-cis/trans-isomerases (Fischer et al., 1984) supports this notion. It has been shown that these enzymes catalyse the cis/trans isomerization of Xaa-Pro bonds, but not of Xaa-nonPro bonds (Scholz et al., 1998a). Recently, a ribosome-associated prolyl isomerase named trigger factor has been described (Stoller et al., 1995) which binds unfolded proteins that do not contain proline residues (Scholz et al., 1998b). However, isomerization does not take place (Scholz et al., 1998a) and it remains unclear, what happens to the non-Pro cis peptide bonds during the course of folding.
The occurrence of non-Pro cis peptide bonds has been associated
with steric strain in proteins (Herzberg
and Moult, 1991) similar to the occurrence of residues with
unfavorable phi/psi-angles and it has been speculated that the location
of these cis peptide bonds is often a peculiar one with respect
to the function of the molecule (Stoddard
et al., 1998; Weiss
et al., 1998). It has been discussed that these sites of strain are
some kind of energy reservoir for the protein. In the course of a
chemical reaction or a conformational change the energy that could be
liberated by a conversion of a cis peptide bond to the trans
conformation could help drive the reaction towards the product. This
notion, however, is speculative at the time and has to await further
experimental confirmation.
The Database
Source: Brookhaven PDB
Data set: non-redundant set of 571 proteins
selected using following criteria:
1.) model structures, incomlete entries were excluded
2.) only structures from X-ray crystallographic data with a
minimum
resolution of 3.5 A were accepted
3.) the maximum amino acid identity between any two protein
chains
of the set was 25%
25% database
Amino Acid Distribution in Proteins| amino acid | 25% database
number % |
from sequence a) % |
| Gly | 12160 7.9 | 7.2 |
| Ala | 13120 8.6 | 8.3 |
| Val | 10507 6.9 | 6.6 |
| Leu | 13264 8.6 | 9.0 |
| Ile | 8724 5.7 | 5.2 |
| Phe | 6265 4.2 | 3.9 |
| Tyr | 5690 3.8 | 3.2 |
| Trp | 2313 1.5 | 1.3 |
| Pro | 7255 4.8 | 5.1 |
| Cys | 2080 1.4 | 1.7 |
| Met | 3267 2.2 | 2.4 |
| Ser | 9176 6.0 | 6.9 |
| Thr | 8940 5.9 | 5.8 |
| Lys | 8533 5.7 | 5.7 |
| Arg | 7310 4.8 | 5.7 |
| His | 3405 2.3 | 2.2 |
| Asp | 9114 5.9 | 5.3 |
| Glu | 9405 6.1 | 6.2 |
| Asn | 7028 4.7 | 4.4 |
| Gln | 5653 3.7 | 4.0 |
a) from primary
structure of 1021 unrelated proteins of known sequence
[P.McCaldon
and P.Argos, Proteins 4, 1988, 99-122]
The 25% database contains 571 protein structures with 153209
peptide bonds. The amino acid
composition of this database agree with that derived from sequence data
(correlation coefficient
between the two sets of numbers is 0.98). The 25% database is
representative and hat is therefore
forms a solid basis for statistical analysis.
Cis Peptide Bonds in Proteins - a General StatisticAll |
<2.0 Å |
2.0 Å - 2.5 Å |
2.5 - 3.5 Å |
|
| Number of Proteins | 571 |
291 |
184 |
96 |
| Number of peptide bonds | 153209 |
72567 |
52194 |
28448 |
| Xaa-Pro | 7413 |
3407 |
2566 |
1440 |
| Xaa-non Pro | 145796 |
69160 |
49628 |
27008 |
| cis peptide bonds | 429 |
232 |
142 |
55 |
(0.28%) |
(0.32%) |
(0.27%) |
(0.19%) |
|
| Xaa-Pro | 386 |
205 |
129 |
52 |
(5.21%) |
(6.02%) |
(5.03%) |
(3.61%) |
|
| Xaa-non Pro | 43 |
27 |
13 |
3 |
(0.029%) |
(0.039%) |
(0.026%) |
(0.011%) |
4.8%
Xaa-Pro (4.7% Stewart
et al. J.Mol.Biol. 214(1990)253)
95.2% Xaa-non
Pro
0.3% of all peptide bonds found to be in cis
90%
Xaa-Pro (about 5% of the total)
10%
Xaa-non Pro (about 0.03 % of the total)
At high resolution, the number of Xaa-Pro cis peptide bonds is
about twice as high than at medium
and low resolution and the number of Xaa-non Pro bonds in cis
conformation is about four times
as high.
Non-proline Cis Peptide Bonds - Distribution in the Database| peptide bond | total number of occurences |
number in cis conformation |
frequency [%] |
| Xaa-aliphatic (except Pro) Xaa-polar Xaa-aromatic all |
53629 74494 17673 145796 |
8 22 13 43 |
0.015 0.030 0.074 0.029 |
| aliphatic-non-Pro polar-non-Pro aromatic-non-Pro all |
58203 70906 16687 145796 |
17 16 10 43 |
0.029 0.023 0.060 0.029 |
| aliphatic-Pro polar-Pro aromatic-Pro all |
2860 3586 967 7413 |
152 157 77 386 |
5.31 4.38 7.96 5.21 |
The 20 amino acids were subdivided into three groups
aliphatic: Ala, Gly, Leu, Ile, Met, Pro, Val
polar : Arg, Asn, Asp, Cys,
Gln, Glu, Lys, Ser, Thr
aromatic: His, Phe, Tyr, Trp
Geometry Geometry of Cis Peptide Bonds in Proteins| Bond | Value [Å]1 | Value [Å]2 | Value [Å]3 | Value [Å]5 | ||||
| N-C |
1.458 | 0.021 | 1.458 | 0.019 | 1.488 | 0.008 | 1.4606 | 0.019 |
| C |
1.527 | 0.017 | 1.525 | 0.021 | 1.508 | 0.009 | 1.527 | 0.025 |
| C-O | 1.236 | 0.016 | 1.231 | 0.020 | 1.244 | 0.008 | 1.238 | 0.013 |
| C-N+ | 1.329 | 0.016 | 1.329 | 0.014 | 1.376 | 0.007 | 1.336 | 0.010 |
| N+-C |
1.456 | 0.013 | 1.458 | 0.019 | 1.457 | 0.009 | 1.459 | 0.007 |
| Angle | Value |
Value |
Value |
Value |
||||
| N-C |
109.2 | 4.0 | 111.2 | 2.8 | 106.7 | 0.6 | 108.66 | 3.0 |
| C |
119.1 | 2.9 | 120.8 | 1.7 | 121.1 | 0.5 | 119.7 | 1.3 |
| C |
120.3 | 5.5 | 116.2 | 2.0 | 118.3 | 0.6 | 119.7 | 1.2 |
| C-N+-C |
126.8 | 4.9 | 121.7 | 1.8 | 127.5 | 0.6 | 127.8 | 1.4 |
| O-C-N+ | 120.3 | 4.3 | 123.0 | 1.6 | 120.3 | 0.6 | 120.6 | 1.1 |
| N+-C |
109.9 | 4.3 | 111.2 | 2.8 | 114.5 | 0.6 | 112.9 | 2.6 |
1 from 27 non-Pro cis peptide bonds in proteins
determined at a resolution of ![]() 2.0 Å
2.0 Å
2 Engh R.A & Huber, R. Acta Cryst.
1991, A47, 392-400
3 from Ala-Asp cis peptide bond in 0.94 Å ConA
structure [Deacon et al., J. Chem. Soc.
Faraday Trans. 1997, 93, 4305-4312]
4 Experimental standard deviations from restrained
refinement
(Dr. A. Deacon and Prof. J. Helliwell, personal
communication.)
5 from 16 small molecule entries retrieved from the Cambridge Structural Database.
6 based on 7 data points only, since not all the amide bonds
from the CSD are in peptides.
Proteins Containing Non-proline Cis Peptide Bonds| PDB Code | Protein | Resolution1 | Non-Proline Cis Peptide Bonds2 | Additional Cis Peptidyl-Prolyl Bonds | Author(s)1 |
| 1AMP | aminopeptidase | 1.8 | D117-D118 | Chevrier et al. | |
| 1BMF | mitochondrial F1-ATPase | 2.85 | D269-D270 (A) | + | Abrahams et al. |
| D256-N257 (D) | |||||
| 1CEC | endoglucanase CelC | 2.15 | W313-N314 | + | Alzari & Dominguez |
| 1CLC | endoglucanase CelD | 1.9 | D177-A178 | + | Alzari & Lascombe |
| 1CTN | chitinase A | 2.3 | G190-F191 | Perrakis et al. | |
| E315-F316 | |||||
| W539-E540 | |||||
| 1DYR | dihydrofolate reductase ( P. carinii) | 1.86 | G124-G125 | + | Champness et al. |
| 1ECE | endocellulase E1 | 2.4 | W319-S320 | + | Sakon et al. |
| 1F13 | coagulation factor XIII | 2.1 | R310-Y311 | + | Weiss & Hilgenfeld |
| Q425-F426 | |||||
| 1GAI | glucoamylase-II | 1.7 | G23-A24 | + | Aleshin et al. |
| 1GHR | 1,3-1,4- |
2.2 | F275-A276 | + | Varghese & Garrett |
| 1GSA | glutathione synthetase | 2.0 | V113-N114 | + | Hara et al. |
| 1HGX | H-G-X phosphoribosyltransferase | 1.9 | L46-T47 | Somoza et al. | |
| 1JAP | matrix-metalloproteinase-8 | 1.82 | N188-Y189 | Bode et al. | |
| 1JPC | snowdrop lectin (mannose-specific) | 2.0 | G98-T99 | Wright & Hester | |
| 1LEN | lentil lectin | 1.8 | A80-D81 | Van Overberge et al. | |
| 1LUC | bacterial luciferase | 1.50 | A74-A75 | + | Fisher & Rayment |
| 1MHL | myeloperoxidase | 2.25 | N549-N550 (C) | + | Fenna et al. |
| 1MKA | 2.0 | P31-N32 | Leesong | ||
| H70-F71 | |||||
| 1NAR | narbonin | 1.8 | G38-F39 | + | Hennig et al. |
| W261-N262 | |||||
| 1NBA | N-carbamoylsarcosine amidohydrolase | 2.0 | A172-T173 | Romao et al. | |
| 1ORO | orotate phosphoribosyltransferase | 2.4 | A71-Y72 | Henriksen et al. | |
| 1PBG | 6-phospho- |
2.3 | W421-S422 | + | Wiesmann & Schulz |
| 1PGS | N-glycosidase F | 1.8 | C204-A205 | + | Norris et al. |
| 1TPL | tyrosine phenol-lyase | 2.3 | V182-T183 | + | Antson et al. |
| 1XYZ | endo-1,4- |
1.4 | H596-T597 | Alzari et al. | |
| 1ZQA | DNA polymerase |
2.7 | G274-S275 | Pelletier & Sawaya | |
| 2CTC | carboxypeptidase A | 1.4 | S197-Y198 | Teplyakov et al. | |
| P205-Y206 | |||||
| R272-D273 | |||||
| 2EBN | endoglycosidase F1 | 2.0 | F45-S46 | + | Van Roey |
| 2HVM | hevamine | 1.80 | A31-F32 | + | Van Scheltinga et al. |
| W255-S256 | |||||
| 2MAD | methylamine dehydrogenase | 2.25 | K129-A130 | Huizinga et al. | |
| 2REB | Rec A protein | 2.3 | D144-S145 | Story & Steitz | |
| 2TMD | trimethylamine dehydrogenase | 2.4 | T70-H71 | Mathews et al. | |
| 3DFR | dihydrofolate reductase ( L. casei) | 1.7 | G98-G99 | + | Filman et al. |
| 4AAH | methanol dehydrogenase | 2.4 | K269-W270 | + | Mathews & Xia |
1 The resolution and the authors quoted are the ones that
appear in the respective PDB entries.
2 In case there are different polypeptide chains in the
coordinate entry, the identifier of the
chain containing the cis peptide bond is given in
parentheses. Cases with non-crystallographic
symmetry are only listed once and are not identified
explicitly.
Non-proline Cis Peptide Bonds and Functional Implications| metal binding | |||
| 1AMP | Aminopeptidase | Asp-Asp | Zn++ |
| dimerization site | |||
| 1BMF | F1 ATPASE | Asp-Asp | |
| 1HGX | Phosphoribosyltransferase | Leu-Thr | |
| 1MKA | Thiol ester hydrolase | His-Phe | |
| 1ORO | Phosphoribosyltransferase (Orotate) | Ala-Tyr | |
| 2TMD | Trimethylamine dehydrogenase | Thr-His | |
| active site | |||
| 1CTN | Chitinase A | Gly-Phe | |
| Glu-Phe | |||
| Trp-Glu | |||
| 1LEN | Lectin | Ala-Asp | |
| 1TPL | Tyrosine phenol-lyase | Val-Thr | |
| 2EBN | Endo-beta-N-acetyl-glucosamidase | Phe-Ser | |
| 3BLM | Beta-lactamase | Glu-Ile | |
| 5CPA | Carboxypeptidase | Ser-Tyr | |
| Arg-Asp | |||
| cofactor/substrate binding | |||
| 1DYR | Dihydrofolatereductase | Gly-Gly | NADPH |
| 1ECE | Endocellulase E1 | Trp-Ser | Cellotetraose |
| 1LLO | Hevamine | Ala-Phe | Allosamidin/ |
| Trp-Ser | Allosamizalon | ||
| 1MKA | Thiolester hydrolase | His-Phe | 3-Decynoyl-N-Acetyl- |
| Cysteamine | |||
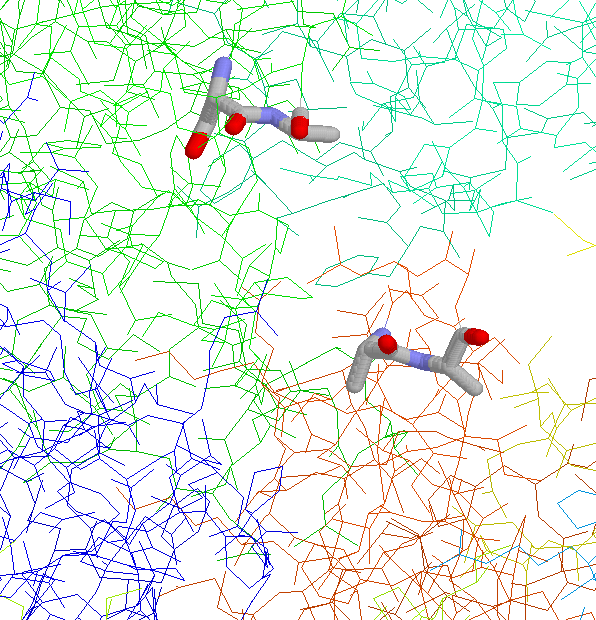
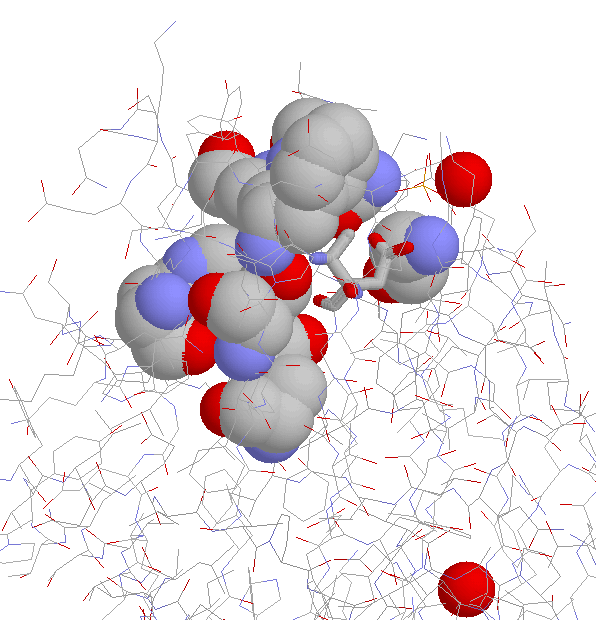
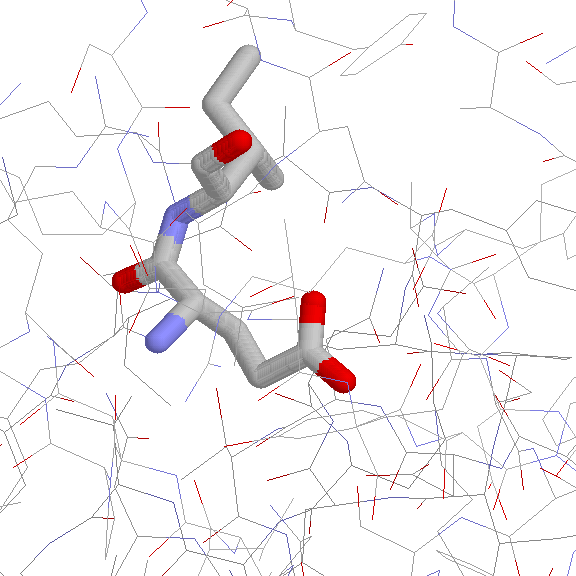
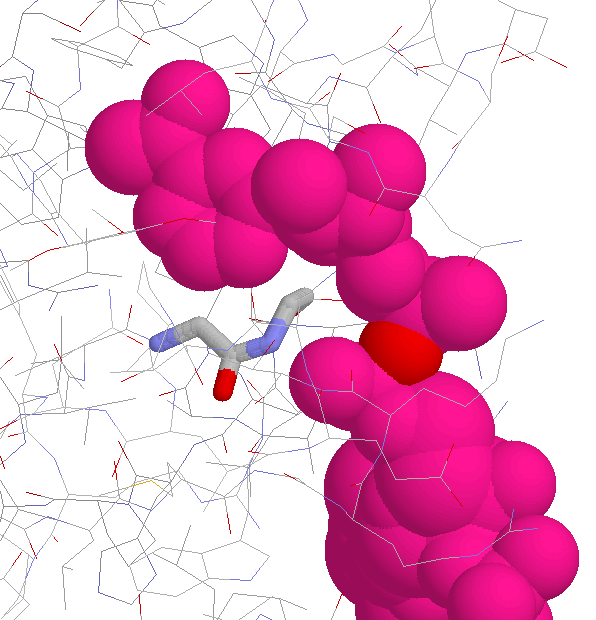
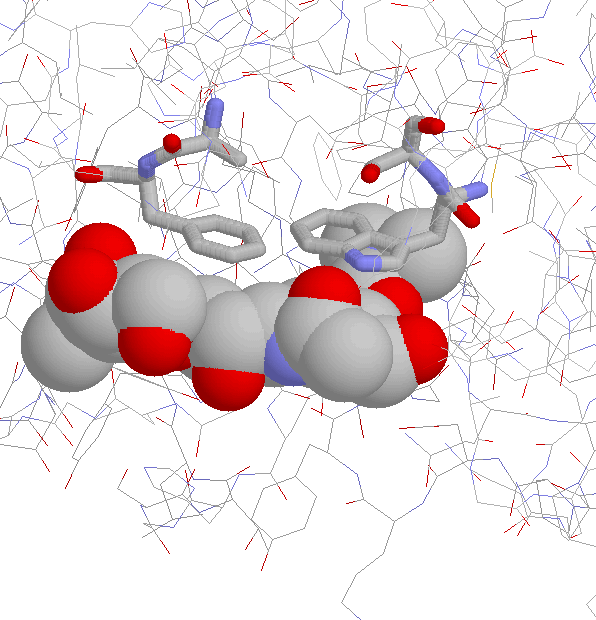
Side Chain Interaction in Non-proline Cis Peptide Bonds
aromatic residue N-terminal: 1CEC, 1CTN, 1ECE, 1GHR, 1NAR, 1PBG
2EBN, 2HVM
the interaction between the aromatic ring and
the side chain (especially the C(beta) atom)
is well defined (JPEG)
aromatic
residue C-terminal 1CTN, 1JAP, 1NAR, 1ORO, 2CTC, 2HVM
4AAH
the strucural pattern is not well defined
(JPEG)
43 non-proline cis peptide bonds were found in the 25% database
10
non-proline cis peptide bonds with interaction between aromatic
an aliphatic
side chains
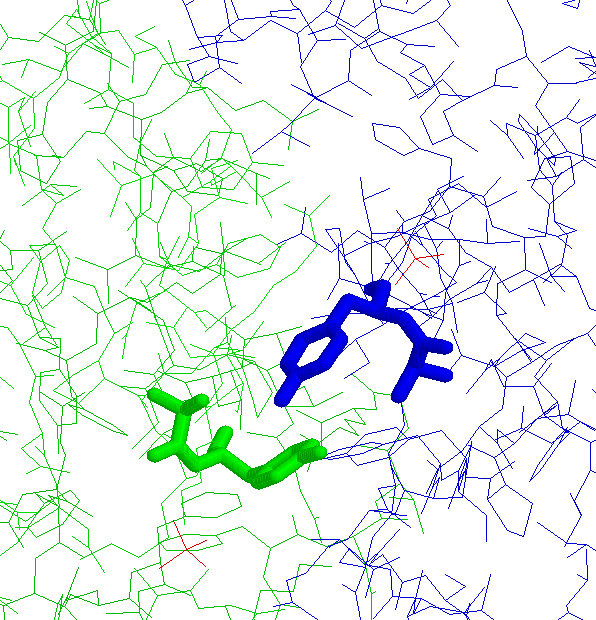
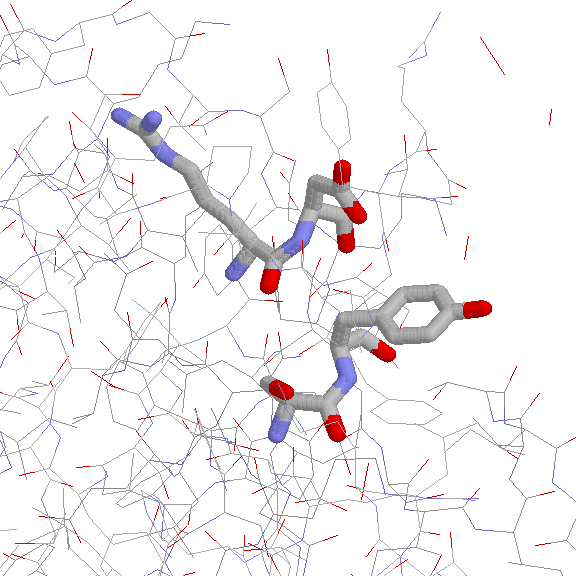
6 non-proline cis peptide bonds with an N-terminal Trp residue
3 Trp-Ser cis peptide bonds in different proteins having
different functions
with identical conformations (JPEG)
Conclusions
Further Reading
|
|
|
|
Beutenbergstraße 11 D-07745 Jena • Germany |
Phone: +49 3641 65-6000 Fax: +49 3641 65-6351 |
E-mail: info@leibniz-fli.de www.leibniz-fli.de |
Data Privacy Imprint |
 |

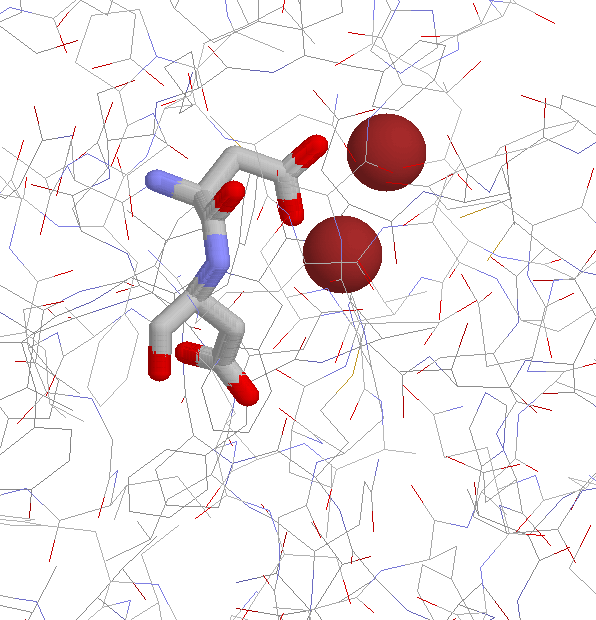{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}