| Three-Dimensional Structures of Peptaibols |
Analysis of the
Peptaibol Bending Geometry
Methods
We describe here a uniform method for the bending geometry analysis of peptaibols that can also be applied to other bent peptide or protein structures
(see also: Kronen M, Goerls H, Ngyuen HH, Reissmann S, Bohl M, Suehnel J , Graefe U., J. Peptide Sci. 2003, 9, 729-744. Crystal structure and conformational analysis of ampullosporin A).
In a first step, we have generated a unique 3D curvilinear axis for all peptaibols using the approach implemented in the program
This axis describes the general folding pattern of a peptide or protein. It can be determined for any peptide or protein backbone independent of its secondary structure characteristics. Therefore, we avoid to use the term helical axis for the 3D curvilinear axis even though for a helical structure it corresponds to the helix axis. In this approach each peptide bond is represented by a local coordinate system with its origin at a characteristic peptide-bond base point (P point). The angle difference between the local axis vectors provides information on the change of direction of the 3D curvilinear axis in passing from peptide group n to n+1.
We use this local bend angle (angle Theta in P-Curves output) to define an N-terminal straight part of the peptaibol structures by assuming that it is characterized by consecutive local bend angles smaller than 9°. In almost all cases straight N-terminal structure parts are observed.a) The length of the straight structure parts is given in the Table. Note, that the first N-terminal amino acid is always counted as 1 and that this numbering scheme may differ from the one used in PDB files.
| Name | Sequence Length | Method of Structure Determination | Straight Structure Parts Beginning at the N-Terminus Used for Determining the Straight-line Axis | Database Code (Database) |
| Alamethicin | 20 | X-ray (3 chains) | chain A: AIB 01 - GLY
11 (PDB numbering: AIB 02 - GLY 12) chain B: AIB 01 - LEU 12 (PDB numbering: AIB 02 - LEU 13) chain C: AIB 01 - AIB 08 (PDB numbering: AIB 02 - AIB 09) |
1AMT (PDB) |
| Ampullosporin A | 15 | X-ray | TRP 01 - AIB 10 | AMPA (CSD) |
| Antiamoebin I | 16 | X-ray (2 chains) | chain A: PHE 01 - LEU
07 (PDB numbering: PHE 02 - LEU 08) chain B: large theta angles at the N-terminus a) |
1JOH (PDB) |
| Antiamoebin I | 16 | X-ray | PHE 01 - LEU 07 | FEJQOA(CSD) |
| Chrysopermin C | 19 | NMR (average structure) | PHE 01 - AIB 10 a) (PDB numbering: PHE 02 - AIB 11) |
1EE7 (PDB) |
| Trichotoxin_A50E | 18 | X-ray (two chains) | chain A: AIB 01 - ALA
10 (PDB numbering: AIB 01 - ALA 10) chain B: AIB 01 - ALA 10 (PDB numbering: AIB 01 - ALA 10) |
1M24 (PDB) |
| Zervamicin IIb | 16 | NMR (20 models); isotropic solvent | TRP 01- AIB 7 (for
all models) (PDB numbering: TRP 02 - AIB 08) |
1DLZ (PDB) |
| Zervamicin IIb | 16 | NMR (20 models); bound to DPC micelles | TRP 01 - AIB 7 (for
all models) (PDB numbering: TRP 02 - AIB 08) |
1IH9 (PDB) |
| Leu1-Zervamicin | 16 | X-ray | Phe 01 - LEU 7 | KIYPUD (CSD) |
a) One exception is
chain B of antiamoebin I (1JOH) with three successive theta angles >
15° at the N-terminus. The other case is found with a theta angle
of 11.5° at the AIB02/SER03 step in chrysospermin C (1EE7).
Whereas the first example is not taken into account in our analysis,
the latter one has been included. As can be seen below the single theta
angle slighty above the limit value of 9° does not prevent the
definition of an apprximately linear N-terminal part in the
chrysospermin structure.
For these approximately
straight N-terminal partial structures we have determined the best straight-line
axis again using P-Curves. Note, that the straight-line axis is
dependent on deviations from the ideal straight geometry seen in the
real structures. For example, in some NMR structures the first
N-terminal amino acids deviate more from the straight-line axis
than the next ones. This affects the orientation of this axis and thus
the extent of bending. Finally, the straight-line axis and the straight part of the 3D curvilinear axis are superimposed.
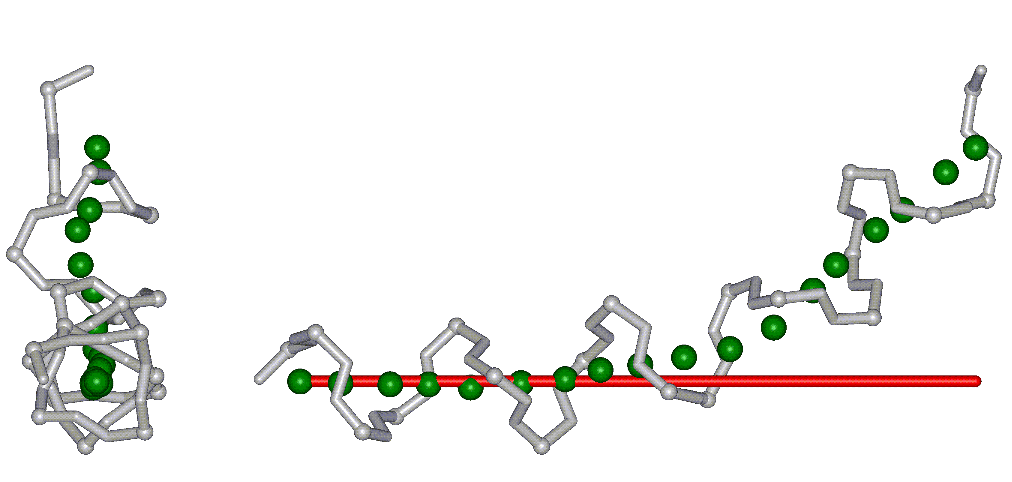
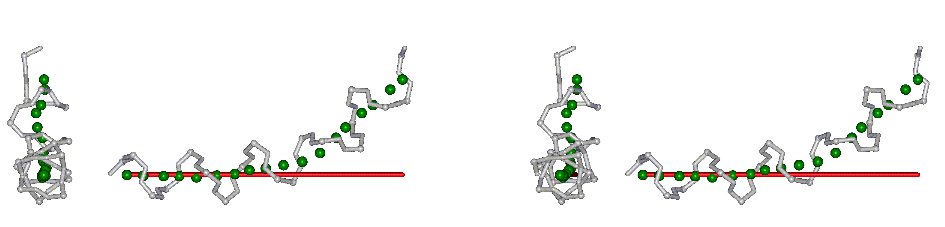
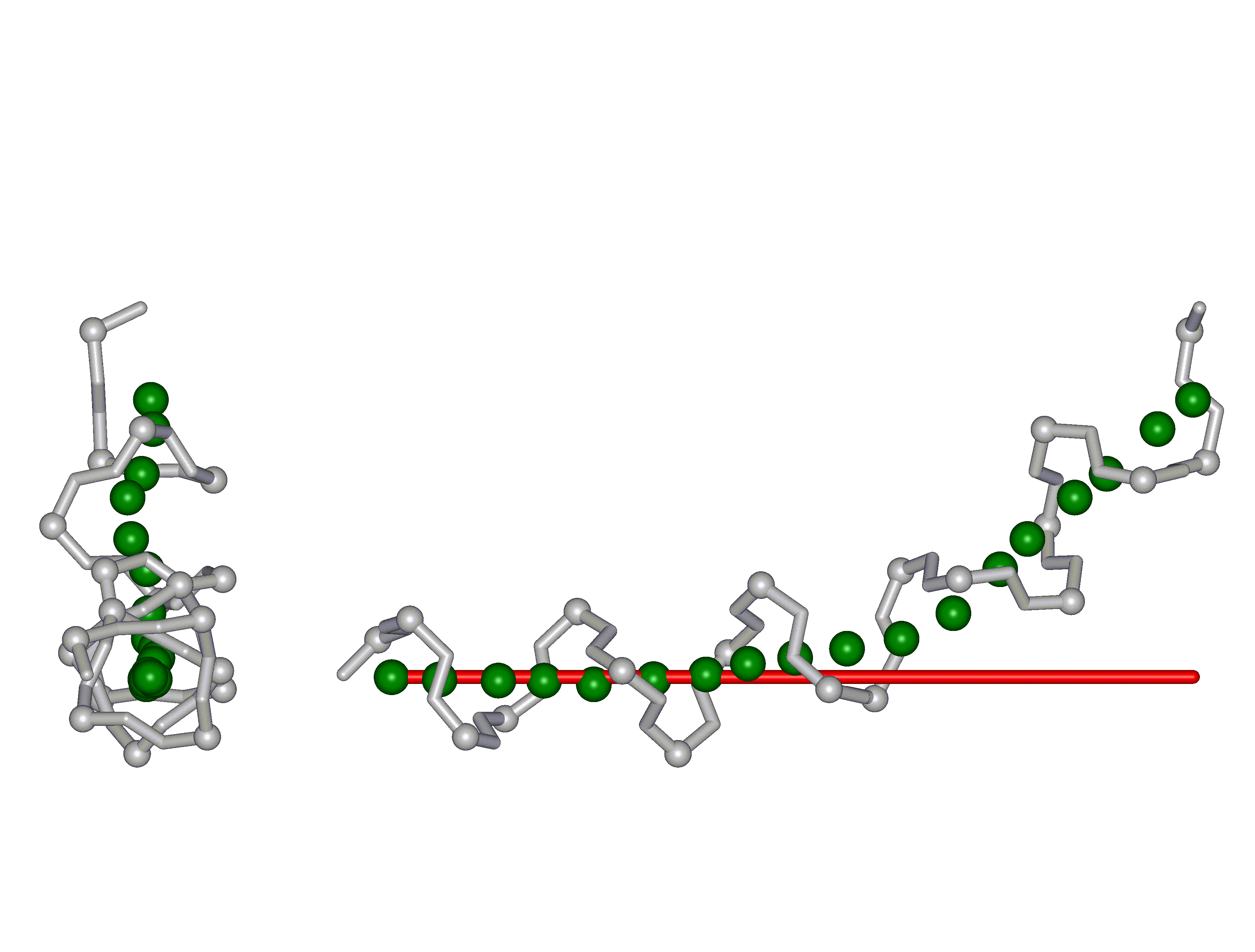
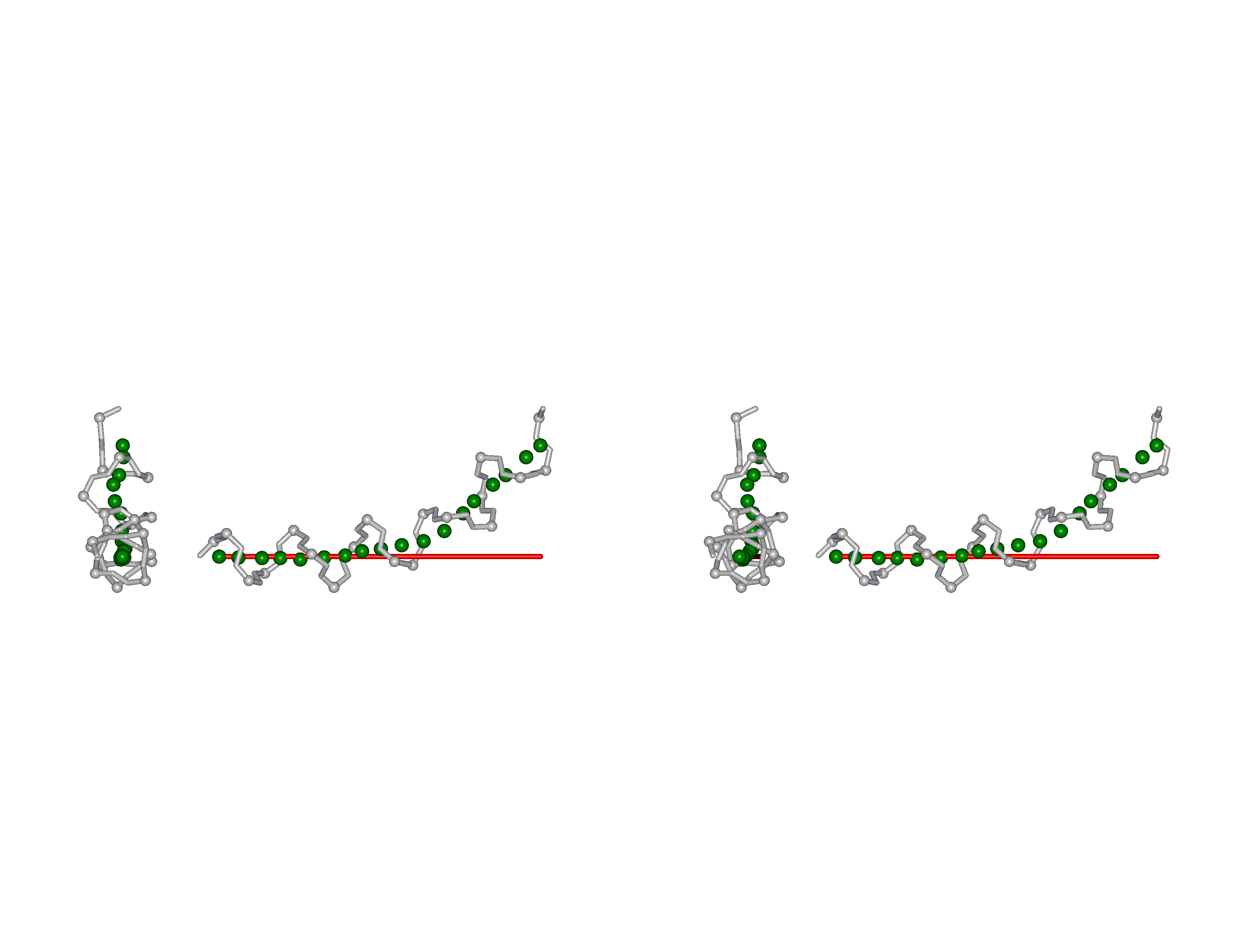
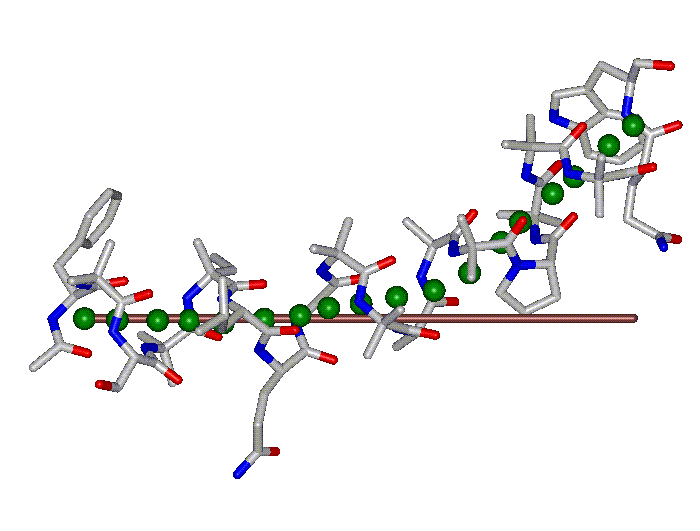
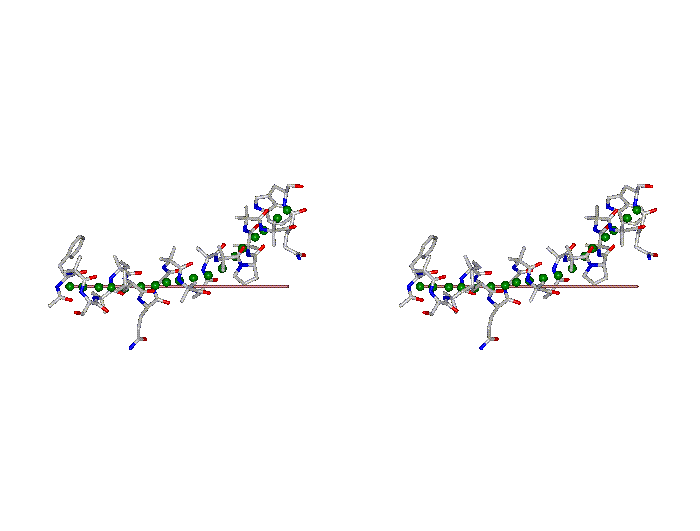
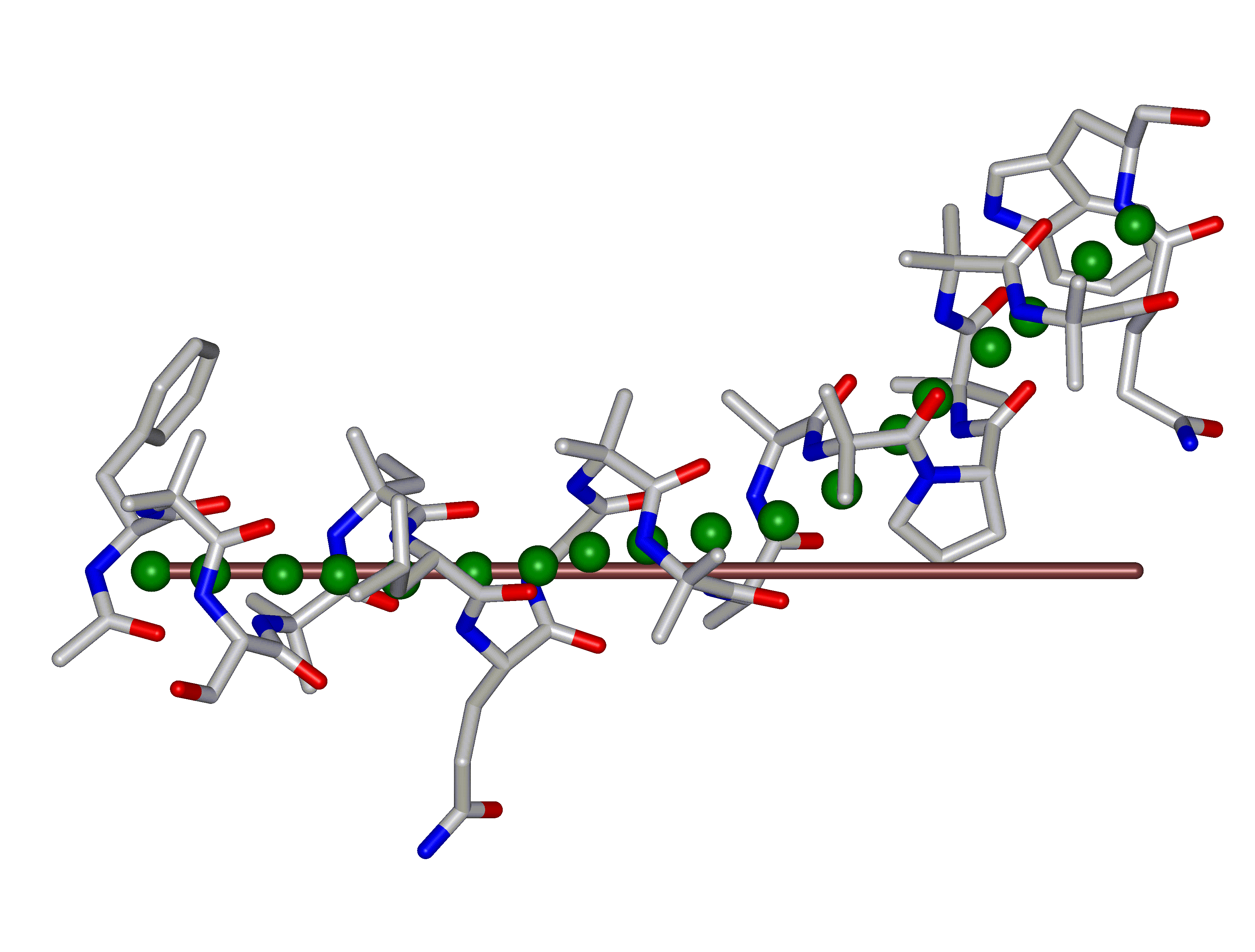
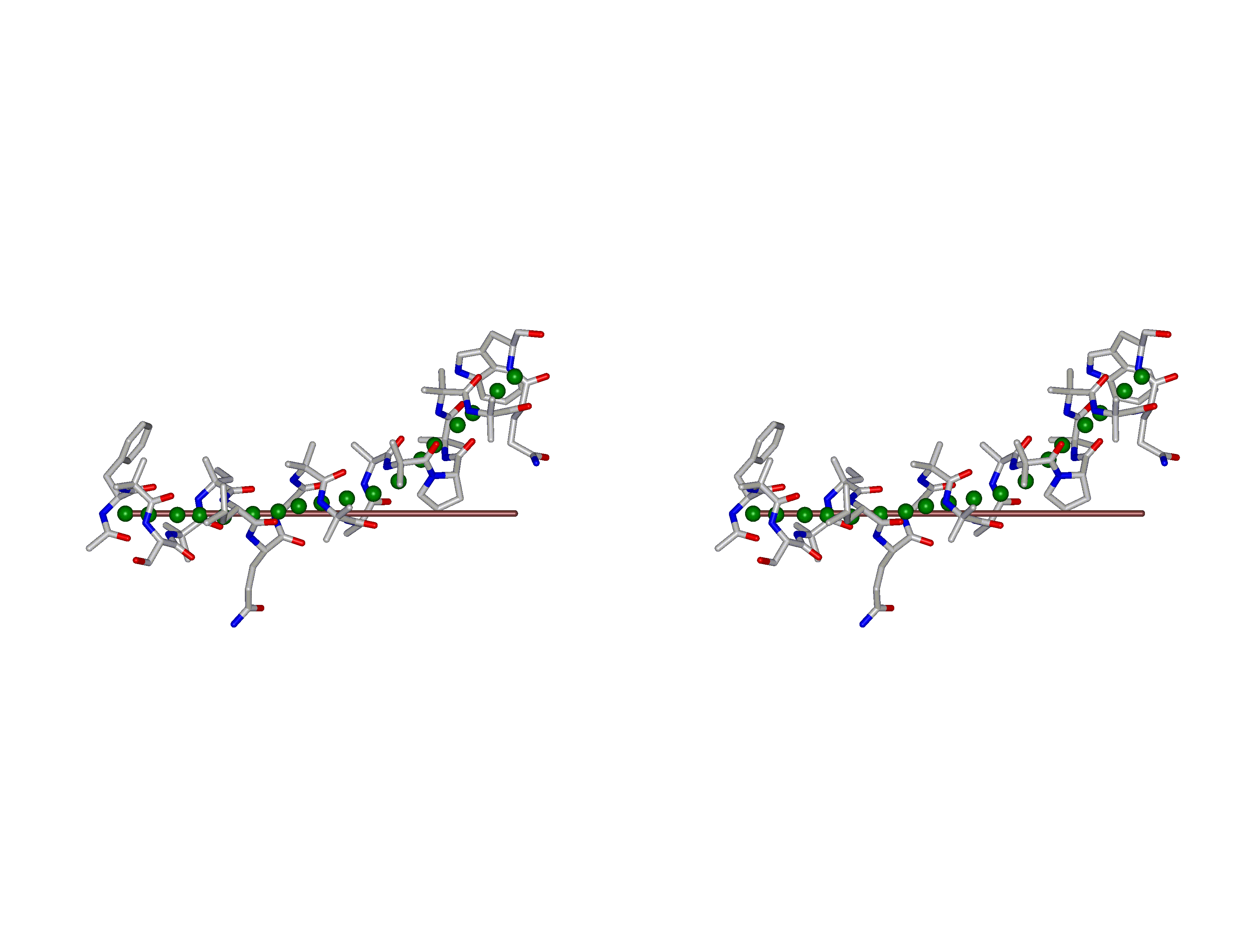
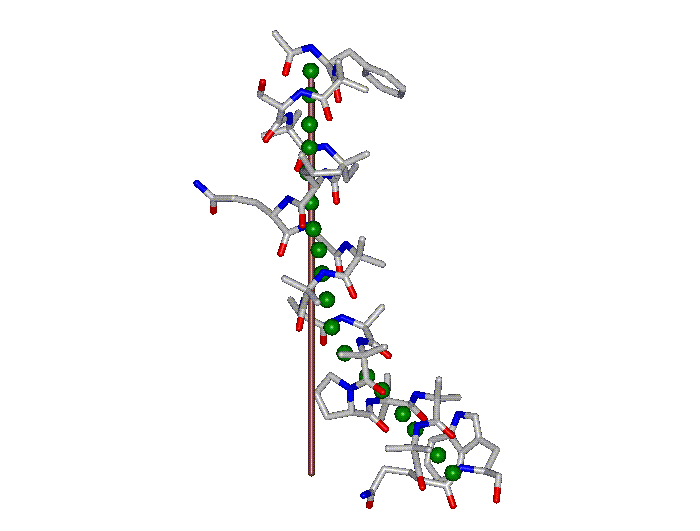
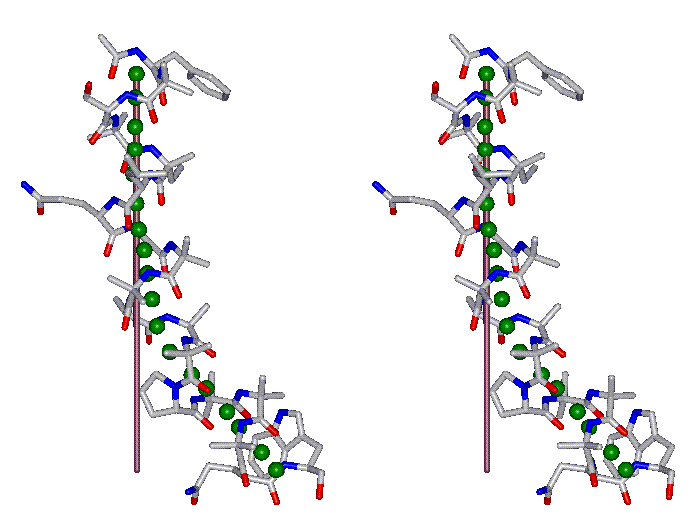
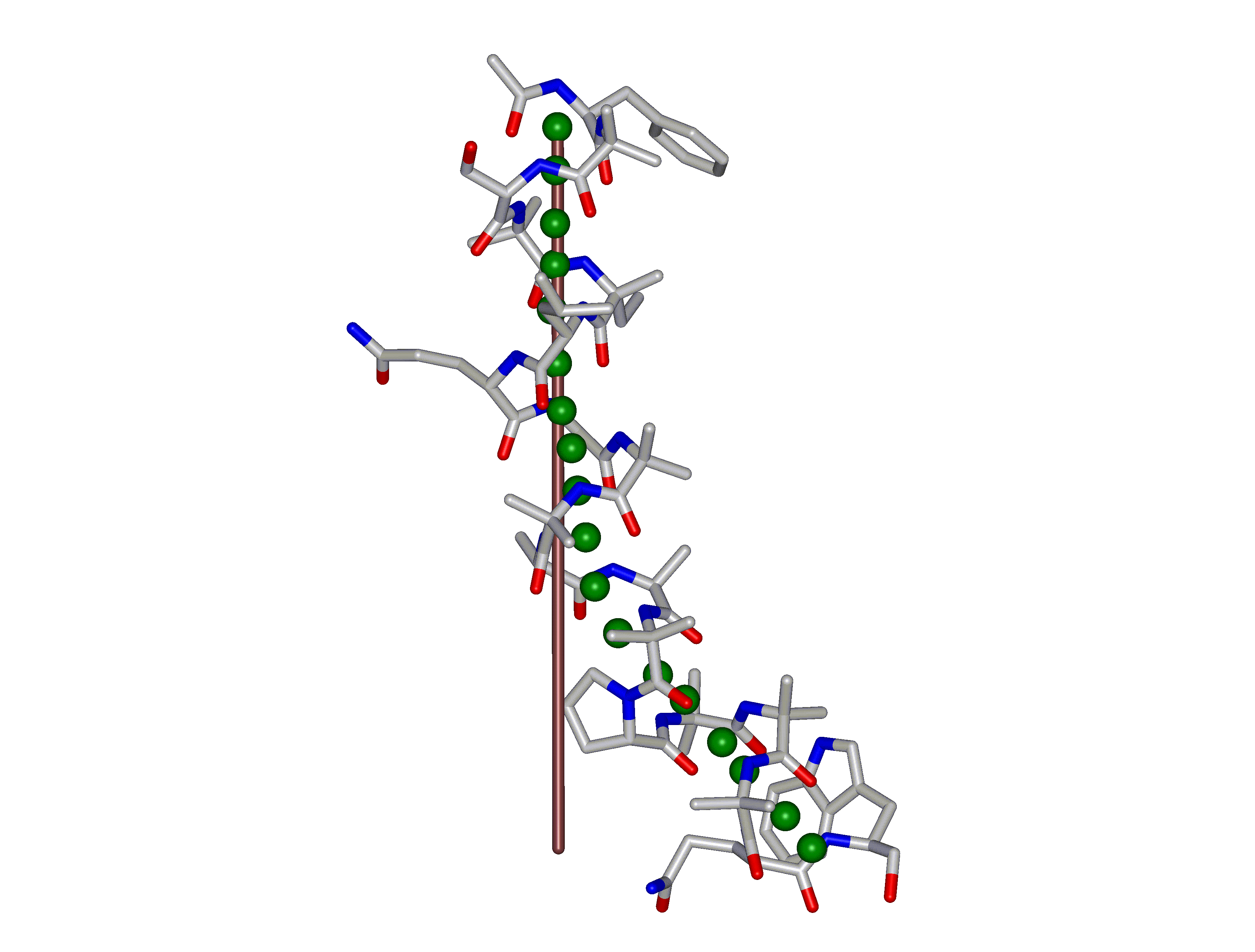
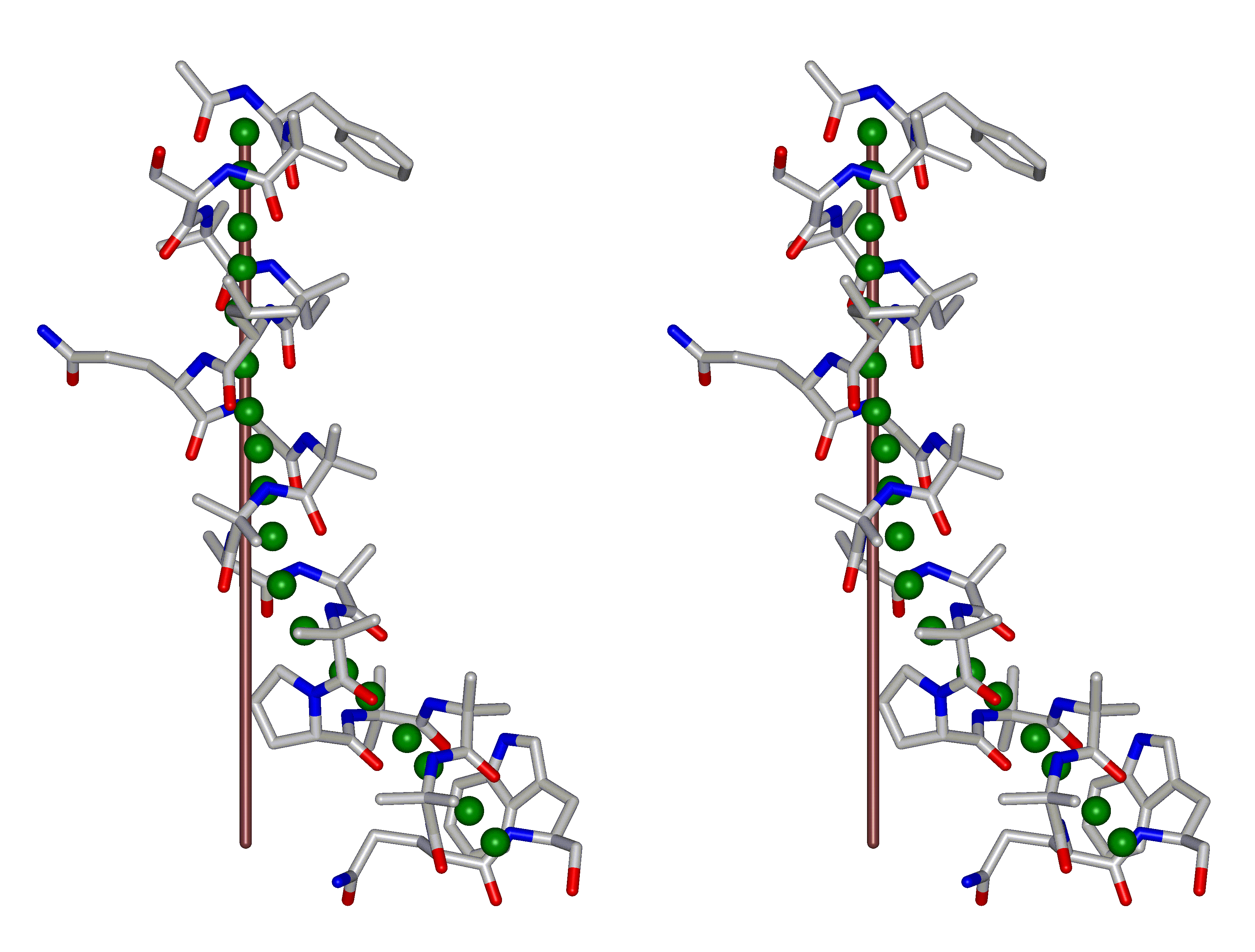
This superposition of the 3D curvilinear and straight-line axes together with either the backbone or the complete structure is shown in the following two Figures for chrysospermin C (1EE7):
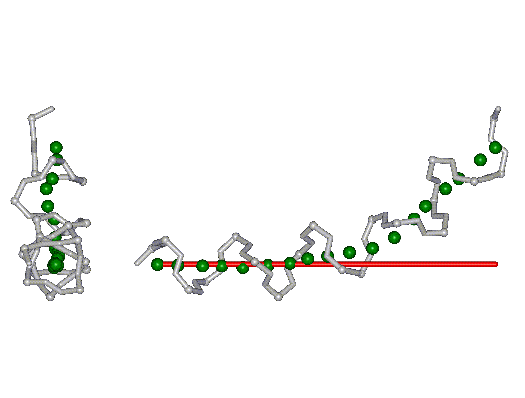
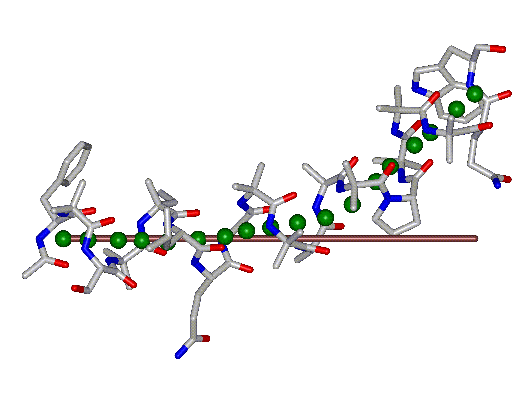
The top view shows that the deviation of the 3D curvilinear axis from a plane is not too large.
In a final step the perpendicular distance of the P points on the 3D curvilinear axis to the straight-line axis (y co-ordinate) and the displacement of the P point projections on the straight-line axis relative to the projection of the first N-terminal base point (x co-ordinate) have been determined using a SYBYL script. These data can be displayed in a two-dimensional 2D bending graph, thereby displaying the full 3D structural information in two dimensions.
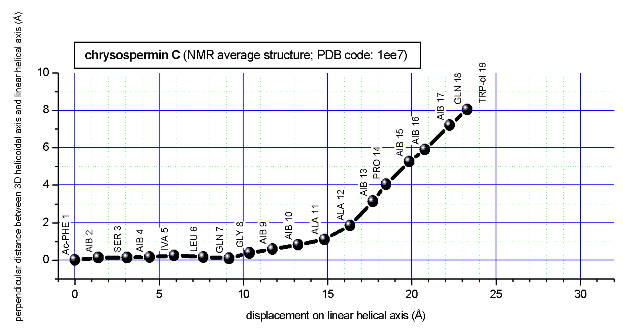
In this graph the axis (x, y=0) corresponds to the straight-line axis of the peptaibol structure. Given two parts of the structure can be approximately described by a straight-line axis an approximate bending angle between these parts can be determined. As can be seen from a comparative bending analysis of peptaibol structures and from the 2D bending graphs of individual structures the C-terminal part of the peptaibol structure is curved in some cases. If one nevertheless tries to place a straight line on the C-terminal part this leaves much room for subjectivity and thus only an angle range can be determined. Even for structures such as chrysospermin C, where the C-terminus appears to be relatively straight one can select different base points for drawing a line through them or, if one does a fit, one can select different structure ranges (ALA 11 - TRP-ol 19 or ALA 12 - TRP-ol 19 ). More detailed information on the bending angle determination can be found here.
Input and output files
These output MOL2 files can be visualized with molecular graphics programs. Tested programs include:
Note that some programs do not correctly display non-natural amino acids.
The SYBYL script can be obtained upon request; contact either Martin Bohl at Tripos, Inc. (mbohl@tripos.com) or Jürgen Sühnel at the FLI (jsuehnel@fli-leibniz.de). P-Curves is available from H. Sklenar at the Max Delbrueck Center for Molecular Medicine (sklenar@mdc-berlin.de).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}