![]()
Determination of Secondary Structure in Proteins by Fourier Transform Infrared
Spectroscopy (FTIR)
Introduction
Basic principles of infrared (IR) absorption
Fourier Transform Infrared (FTIR) spectroscopy
Band assignments
Amide vibrations
Amino acid side chain vibrations
Secondary structure - model compounds
Beta sheet structures
Helical structures
alpha helix
310-helix
Turn structures
Secondary structure in proteins
Deconvolution of the amide I band
Second derivative spectra and curve fitting
New trends
Introduction
During the last years the use of Fourier Transform Infrared spectroscopy
(FTIR) to determine the structure of biologicalmacromolecules has dramatically expanded.
The complete three-dimensional structure of a protein at high resolution can be determined by X-ray crystallography.
This technique requires the molecule to form a well ordered crystal which is not possible for all proteins. An alternative to X-ray crystallography is multidimensional nuclear magnetic resonance (NMR) spectroscopy. Using NMR spectroscopy structures of the proteins can be determined in solution. The interpretation of the NMR spectra of large proteins is very complex,
so its present application is limited to small proteins (~15-25 kDa). These limitations have led to the development of alternative methods that are not able to generate structures at atomic resolution but provide also structural information on proteins (especially on secondary structure). These methods include circular dichroism (CD) and vibrational (infrared and RAMAN) spectroscopy.
The new technique of FTIR spectroscopy requires only small amounts of proteins (1mM) in a variety of environments.
Therefore, high quality spectra can be obtained relatively easy without problems of background fluorescence,
light scattering and problems related with the size of the proteins. The omnipresent water absorption can be mathematically
substracted. Methods are now available that can separate subcomponents that overlap in the spectra of proteins. These facts have made practical biological systems amenable to studies by FTIR spectroscopy.
Basic principles of infrared (IR) absorption
We shalllimit ourselvesto a few lines here, because many textbooks present excellent descriptions
of the basis of IR spectroscopy (Campbell & Dwek, in Biological Spectroscoy,Benjamin Cummings,, Menl Park, CA 1984,Brey, Physical Chemistry and its Biological Applications, AcademicPress, NewYork, 1984,p.133).
IR spectroscopy is the measurement of the wavelength and intensity of the absorption of infrared light by a sample. Infrared light is energetic enough to excite molecular vibrations to higher energy levels.

Electromagnetic spectrum
| frequency range (Hz) | wavelength range | type of radiation | type of transition |
| 1020 - 1024 | 10-12 - 10-16 m | gamma rays | nuclear |
| 1017 - 1020 | 1 nm - 1 pm | x-rays | inner electrons |
| 1015 - 1017 | 400 - 1 nm | ultraviolet light | outer electrons |
| 4.3x1014 - 7.5x1014 | 700 - 400 nm | visible light | outer electrons |
| 1012-1014 | 2.5 um - 700 nm | infrared light | vibrations |
| 108 - 1012 | 1 mm - 2.5 um | microwaves | rotations |
| 100 - 108 | 108 - 1 m | radio waves | spin flips |
The infrared spectra usually have sharp features that are characteristic of specific types of molecular vibrations, making the spectrauseful for sample identification.
Table of characteristic IR bands
| X-H vibrations | bond | wavenumbers (cm-1) |
| hydroxyl | O-H | 3610-3640 |
| amines | N-H | 3300-3500 |
| aromatic rings | C-H | 3000-3100 |
| alkenes | C-H | 3020-3080 |
| alkanes | C-H | 2850-2960 |
| triple bonds | 2500-1900 | |
| double bonds | 1900-1500 | |
| deformation/heavy atoms | 1500- |
For a molecule of N atoms, 3N-6 fundamental vibrations (or normal modes) exist (3N-5 if the molecule is linear). For the linearCO2 molecule 4 normal modes to expect.
Normal modes for CO2
| cm-1 | IR | RAMAN | ||
| stretching (sym.) | -> <- O==C==O | 1340 | - | + |
| stretching (asym.) | -> <- <- O==C==O | 2349 | + | - |
| deformation | /| /| O==C==O \| | 667 | + | - |
| deformation | + - + O==C==O | 667 | + | - |
Fourier Transform Infrared (FTIR) spectroscopy
To use the Fourier Transform Infrared Spectroscopy, a continuum source of
light (such as a Nernst Globar) is used to produce light overa broad range of infraredwavelengths. Light coming from this continuum sourceis split into two paths using ahalf-silvered mirror; this light isthen reflected from two mirrorsback onto the beamsplitter, where it is recombined. One of thesemirrors is fixed, and the second is movable.If the distance from the beamsplitterto the fixed mirror is notexactly the same as the distance fromthe beamsplitter to thesecond mirror, then when the two beams are recombined, therewill be a small difference in thephase of the light betweenthese two paths. Because of the "superposition principle" constructiveand destructive interference exist for different wavelengthsdepending of the relative distancesof the two mirrors from the beamsplitter.
It can be shown that if the intensity of light is measured and plotted as a function of the position of the movable mirror, the resultant
graph is the Fourier Transform of the intensity of light as a function of wavenumber . In FTIR spectroscopy , the light is directed onto the sample of interest, and the intensity is measured using an infrared detector. The intensity of light striking the detector is measured as a function of the mirror position, and this is then Fourier-transformed to produce a plot of intensity vs. wavenumber.
As radiation source an Michelson Interferometer is used. Its basic outline is shown in green in the drawing below.
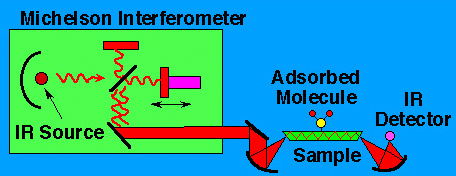
It is necessary to increase the sensitivity somehow, because the absorption due to one monolayer of molecules typically results in a change in intensity of only about one part in 105 .For semiconductors, one way of increasing the sensitivity is touse multiple internal reflection. In thistechnique, the edges of the sample are polished, and the light is sent in at an angle.The light bounces aroundinside the sample, makingabout 30-50 bounces. This increases the sensitivity by about a factor of30-50, making it possible to measure the absorption of less than one monolayer of molecules ona surface.
Band assignments
Amide vibrations
The peptide group, the structural repeat unit of proteins, gives up to 9
characteristic bands named amide A, B, I, II ... VII. The amide A band (about
3500 cm-1) and amide B (about 3100 cm-1) originate from a Fermi resonance between the first overtone ofamide II and and the N-H stretching vibration. Amide I and amide II bands
are two major bands of the proteininfrared spectrum. The amide I band (between 1600 and 1700 cm-1) is mainly associated with the C=O stretchingvibration(70-85%)and isdirectly related to thebackbone conformation. Amide II results from the N-Hbending vibration (40-60%) and from the C-N stretching vibration (18-40%).
This band is conformationalsensitive. Amide III and IV are very complex bands resulting from a mixture
of several coordinatedisplacements. The out-of-plane motions are found in amide V, VI andVIII.
Amide A is with more than 95% due to the the N-H stretching vibration. This mode of
vibration is not depend on the backbone conformation but is very sensitive to the strength of a hydrogen bond (between 3225 and
3280 cm-1 for hydrogen bond length from 2.69 to 2.85 angstrom, (Krimm & Bandekar Adv Protein Chem 1986;38:181-364).
Amide I is the most intense absorption band in proteins. It is primilary goverend by the
stretching vibration of the C=O (70-85%) and C-N groups (10-20%). Its frequency is found in the range between 1600 and 1700
cm-1. The exact band position is determined by the backbone conformation and
the hydrogen bonding pattern.
Amide II is found in the 1510 and 1580 cm-1 region and it is more complex than amide I. Amide II derives mainly from
in-plane N-H bending (40-60% of the potential energy). The rest of the potential energy
arises from the C-N (18-40%) and the C-C (about 10%) stretching vibrations.
Amide III, V are very complex bands dependent on the details of the force field, the nature of side chains and
hydrogen bonding. Therefore these bands are of little use.
Amino acid side chain vibrations
The presence of bands arising from amino acid side chains must be recognized
before attempting to extract structural informationfrom the shapes of amide I and amide II bands. The contribution of the side
chain vibrations in the region between 1800 and 1400 cm-1 (amide I and amide II region) has been thoroughly investigatedby Venyaminov & Kalnin 1990 (Biopolymers 1990;30(13-14):1243-57). Among the 20 proteinogenous amino acids only 9 (Asp, Asn, Glu, Gln, Lys, Arg, Tyr, Phe, His) show a significant absorbance in the region discussed above. The
contribution of the different amino acid side chains were fitted by a sum of Gaussian and Lorentzian components.
| AS | vibration | cm-1 | A0 (l/mol/cm) | FWHH (cm-1) | surface (x10-4 l/mol/cm) | |
| Asp | -COO st as | pH>pK (~4.5) | 1574 | 380 | 44 | 5.5 |
| -COOH st | pH<pK (~4.5) | 1716 | 280 | 50 | 4.1 | |
| Glu | -COO st as | pH>pK (~4.4) | 1560 | 470 | 48 | 7.1 |
| -COOH st | pH<pK (~4.4) | 1712 | 220 | 56 | 3.6 | |
| Arg | -CN3H5+ st as | 1673 | 420 | 40 | 4.3 | |
| st s | 1633 | 300 | 40 | 3.6 | ||
| Lys | -NH3+ bd as | 1629 | 130 | 46 | 1.8 | |
| bd s | 1526 | 100 | 48 | 1.3 | ||
| Asn | -C=O st | 1678 | 310 | 32 | 2.7 | |
| -NH2 bd | 1622 | 160 | 44 | 2.5 | ||
| Gln | -C=O st | 1670 | 360 | 32 | 3.1 | |
| -NH2 bd | 1610 | 220 | 44 | 3.5 | ||
| Tyr | ring-OH | pH<pK (~10) | 1518 | 430 | 8 | 1.0 |
| ring-O | pH>pK (~10) | 1602 | 160 | 14 | 0.7 | |
| 1498 | 700 | 10 | 2.5 | |||
| His | ring | 1596 | 70 | 14 | 0.3 | |
| Phe | ring | 1494 | 80 | 6 | 0.2 | |
| terminal | ||||||
| -COO st as | 1598 | 240 | 47 | 3.5 | ||
| -COOH st | 1740 | 170 | 50 | 2.1 | ||
| -NH3+ bd as | 1631 | 210 | 54 | 3.8 | ||
| bd s | 1515 | 200 | 60 | 4.3 | ||
| -NH2 bd | 1560 | 450 | 46 | 7.5 |
(according to Venyaminov & Kalnin Biopolymers 1990;30(13-14):1243-57)
Secondary structure - peptide model compounds
A large number of synthetic polypeptides has been used for the characterization of infrared spectra for proteins with adefinedsecondary structure content. For example, polylysine adopts an random, beta-sheet or alpha-helical structures in dependence on temperature and pH of the solution (Susi et al. 1967). Experimental and theoretical work on a large number of synthetic polypeptides has provided insights on the variability of the frequencies for each structure. The large amount of data published by Krimm & Bandekar, Adv Protein Chem 1986;38:181-364 gives an insight in the nature of the amide bond.
Beta sheet structures
(beta strand)
The frequencies of the main absorption bands from synthetic polypeptides
adopting an antiparallel chain structure have been compiled by Chirgadze & Nevskaya (Biopolymers 1976 Apr;15(4):637-48). From this follows, that the amide I absorption are essentially determined by the backbone conformation and independent of the amino acid
sequence, its hydrophilic or hydrophobic properties and charge. The average frequency of the main
component is about 1629 cm-1 with a minimum of 1615 cm-1 and a maximum of 1637 cm-1. The average value for the second frequency is 1696 cm-1 (lowest value 1685 cm-1). The parallel beta sheet structure is not common in synthetic polypeptides.
The main band is located near 1640 cm-1
Helical structures
The alpha-helix: For alpha-helical structures the mean frequency was found to be 1652 cm-1
for the amide I and 1548 cm-1 for the amid II absorption (Chirgadze & Nevskaya Biopolymers 1976 Apr;15(4):637-48). The half width of the alpha-helix band depends on the stability of the helix. For the most stable helices, the half-width
of about 15 cm-1 corresponds with a helix-coil transition free energy of more than 300 cal/mole. Other
helices display half-widths of 38 cm-1 and helix-coil transition free energies of about 90 cal/mole.
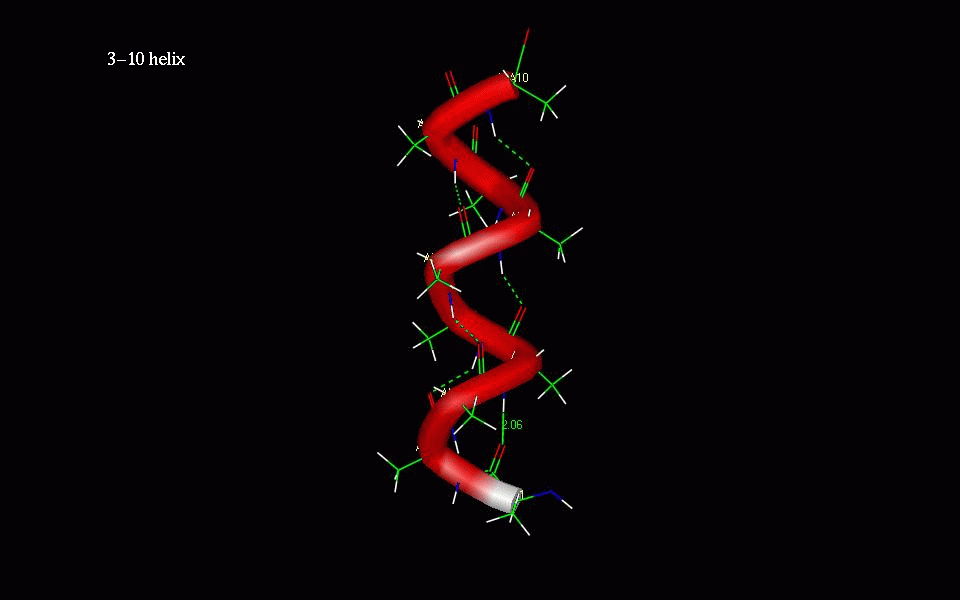
The 310-helix differs from the alpha-helix in that the internal hydrogen bonding occurs
between residues i and i+3 instead of i and i+4 in alpha helices.
Turn structures
The beta turn structure involves 4 amino acid residues which form a loop so that the two chain segments separated by theturn adopt an antiparallel orientation and form a i to i+3 hydrogen bond. A number of turn structures havebeen identified from protein structures: type I (42%, non-helical), type II (15%, non-helical, requires Gly in position 3)and type III (18%, correspondsto one turn of 310 helix) (Krimm & Bandekar Adv Protein Chem 1986;38:181-364). Assignment of beta turns from normal mode analysis in insulin demonstrates a strong overlapping of the four types of beta turn with the alpha-helical absorption (Krimm & Bandekar Adv Protein Chem 1986;38:181-364). An absorption near 1680 cm-1 is now clearly assigned to beta turns.
Secondary structure in proteins
The shape of the amide I band of globular proteins is characteristic of their secondary structure. With a publication byByler & Susi (Biopolymers 1986 Mar;25(3):469-87 ) the determination of secondary structures in proteins from FTIR spectra really started. This was possible by the availability of high signal-to-noise ratio digitalised spectra obtained by the FTIR spectrometer and by the access to computers and software able to perform many operations on the spectra in a short time.
Deconvolution of the amide I band
The concept of Fourier self deconvolution is based on the assumption, that
a spectrum of single bands (each narrow bandis characteristic for a secondary structure) is broadened in the liquid
or solid state. Therefore the bands overlap and cannot be distinguished in the amide envelope. A curve fitting procedure can
be applied to estimate quantitatively the areaof each component representing a type ofsecondary structure. In the pioneer work by Susi & Byler (Methods Enzymol 1986;130:290-311) the amide I was deconvoluted with a Lorentzian line shape function and a resolution enhancement factor
of 2.4 was applied. The deconvoluted spectrum was fitted with Gaussian band shapes by an iterative
curve fitting procedure. The results are in good agreement with with the X-ray crystallographic structures of the proteins.
| a) | b) | ||||||
| sec. structure | Mean (cm-1) | RMS (cm-1) | Max (cm-1) | Mean (cm-1) | RMS (cm-1) | Max (cm-1) | Region (cm-1) |
| turns | 1694 | 1.7 | 2 | - | - | - | |
| 1688 | 1.1 | 2 | - | - | - | ||
| 1683 | 1.5 | 2 | 1678 | 2.1 | 5 | 1682-1662 | |
| 1670 | 1.4 | 2 | 1670 | 2.9 | 5 | ||
| 1663 | 2.2 | 4 | 1664 | 1.0 | 3 | ||
| alpha-helix | 1654 | 1.5 | 3 | 1656 | 1.5 | 3 | |
| 1648 | 1.6 | 3 | 1662-1645 | ||||
| unordered | 1645 | 1.6 | 4 | 1641 | 2.0 | 3 | 1645-1637 |
| beta sheet | 1624 | 2.4 | 4 | 1624 | 2.5 | 5 | |
| 1631 | 2.5 | 3 | 1633 | 2.1 | 4 | 1637-1613 | |
| 1637 | 1.4 | 3 | - | - | - | ||
| 1675 | 2.6 | 4 | 1685 | 2.1 | 4 | 1689-1682 |
Proteins in solution (Susi & Byler (Methods Enzymol 1986;130:290-311), a) ) or as hydrated film on a ATR plate (Goormaghtigh et al. Eur J Biochem 1990 Oct 24;193(2):409-20, b) ); the mean frequency of each component is reported with the root mean square (RMS) and the maximum deviation (Max).
Second derivative spectra and curve fitting
The spectra discussed in this chapter are collected with a IFS 66 spectrometer (Fa. BRUKER). The protein spectra are detected at a resolution of 1.5 cm-1. The native proteins were solved in water (pH 6.5). All proteins were denatured
by dissolving in water and heating up to 95 oC for 50 minutes. The aqueous protein solutions to be lyophilized were frozen in liquid nitrogen. The lyophilized proteins were measured at 1.5 mg protein per 300 mg of KBr. After homogenizing the lyophilized protein
and KBr were pressed into pellets by using a 12-ton hydraulic press.
The author thanks Wilfried Hartmann and Gisela Werner from the BRUKER SAXONIA GMBH for the access to the spectrometer facilities and for the excellent technical assistance.
The resolution-enhanced spectra allow the identification of the various
secondary structures present in the protein. Most of the peakpositions were easily found in thesecond derivative spectra. An example for the second derivative of a Gaussian
and a Lorenztiancurve is shown in this figure (JPEG). Using only the peak positions from the second derivative spectrum, for lysozyme (JPEG), 12 different peaks were found in the amid I region. In addition to the frequency position (a1), information on the width (a2) and the maximum absorption intensity (a0) of the individual bands can be obtained from the
second derivative. However, accurate determination of the bandwidth from the maxima in the second derivative is complicated due to the presence of neighboring peaks. Despite this inaccuracy, the obtained parameters can successfully be used as input parameters for a fitting procedure.
However, the peak positions were sometimes difficult to distinguish. This difficulty
arose when either one of the two peaks showed up as a shoulder instead of a separate peak in the second derivative. In general, the bandwidth of the structural components are in the range of
8-28 cm-1. In some second derivative spectra, peaks appeared with very small band
widths. It was assumed, that these small fluctuations originated from noise. Using the additional parameters from the second derivative spectra for lysozyme (JPEG), the resulting peak number is 6. The Table compares the secondary structure contents for different techniques with results from X-ray crystallography.
| X-ray | FTIR | ||||
| a) | STRIDE b) | a1 c) | a1 | a0,a1,a2 | |
| helix | 45 | 46 | 56 | 41 | 42 |
| beta-sheet | 19 | 17 | 27 | 29 | 16 |
| turn | 23 | 28 | 16 | 16 | 36 |
| random | 13 | 9 | 1 | 14 | 6 |
Using the parameters a0, a1 and a2, the secondary structure content derived
from FTIR spectra is in agreement with X-raycrystallography data. The structural components werequantified by the integrated areas of the respective peaks. This implies
that the effective absorptivitieswere assumed to be equal. This assumption wasvalidatedin Byler & Susi (Biopolymers 1986 Mar;25(3):469-87).
CD spectroscopy is a well established technique for the analysis of secondary structure of proteins in aqueous solution. This technique seems to be less reliable for the study of aggregated proteins, inclusion bodys or membrane bound proteins due to light scattering problems associated with large membrane fragments or aggregates. FTIR spectroscopy has proved to be a powerful tool for investigations on proteins discussed above.
| LDH | FAB | CSC | ||||
| Xray | FTIR | Xray | FTIR | Xray | FTIR | |
| helix | 43 | 49/25 | 49 | 19/6 | 64 | 64/4 |
| beta-sheet | 19 | 21/15 | 14 | 39/18 | 1 | 15/14 |
| turn | 30 | 27/15 | 28 | 33/18 | 23 | 19/31 |
| random | 8 | 3/42 | 9 | 9/39 | 1 | 2/49 |
The FTIR spectra in the amid I region of LDH (lactate dehydrogenase), FAB (fab fragment, mouse antibody), CSC (citrate synthase), LYS (lysozyme) were recorded according to the procedure described in the top of this chapter. The second derivatives of all spectra were calculated using the spectrometer software OPUS. Before starting the fitting procedure, the obtained depths of the minima in the second derivative spectrum and, subsequently, the calculated maximum intensities were corrected for the interference of all neighboring peaks. The curve fitting is performed by stepwise iterative adjustment towards a minimum root-mean-square error of the different parameters determining the shape and position of the absorption peaks.
spectra of native proteins (click to enlarge)
 LYS
LYS LDH
LDH FAB
FAB CSC
CSC
The data reveal that theamide I band for all proteins consists of six orseven major components which were found in all spectra.The helix content derived from the amid I regionfor lysozyme, lactate dehydrogenase and citrate synthase is inagreement with the data from X-raycrystallography. The helix content in the FAB fragment of mouse antibody is tolow,the beta-sheetcontentto high. Visual inspection of the amide I envelope of the native and thermally denaturedstates revealed a striking difference in the band shape between them. For the native state, the band isfairly asymmetric and has a peak maximum around 1650 cm-1 which corresponds to alpha-helical structure. In contrast, thedenatured proteins show an additional maximum between 1620 and 1640 cm-1, indicative of the predominance of beta-sheetand unorderedstructures.
spectra of thermally denatured proteins (click to enlarge)
 LYS
LYS LDH
LDH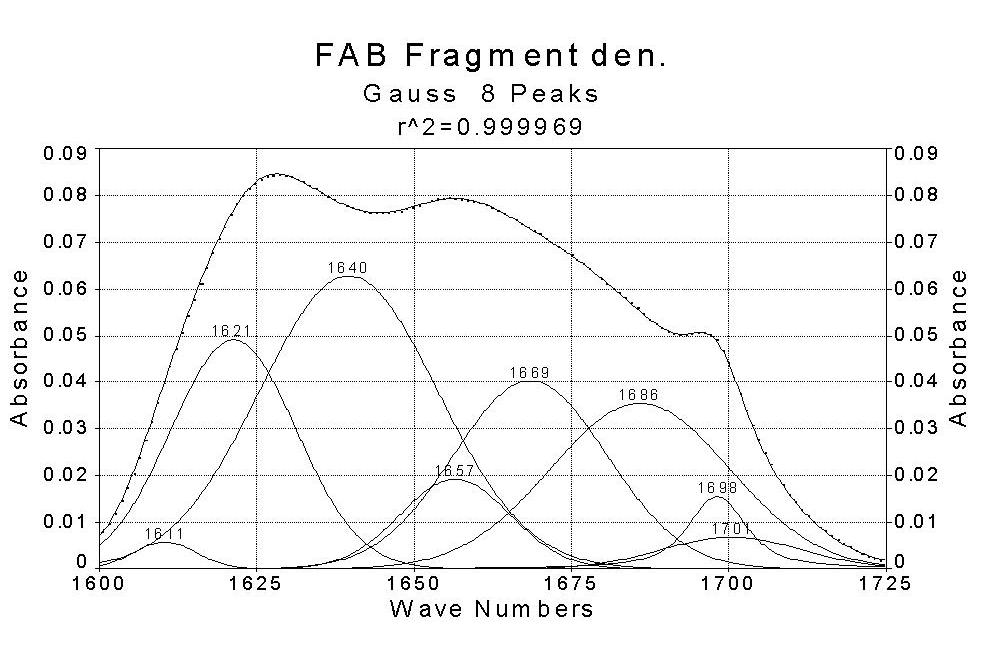 FAB
FAB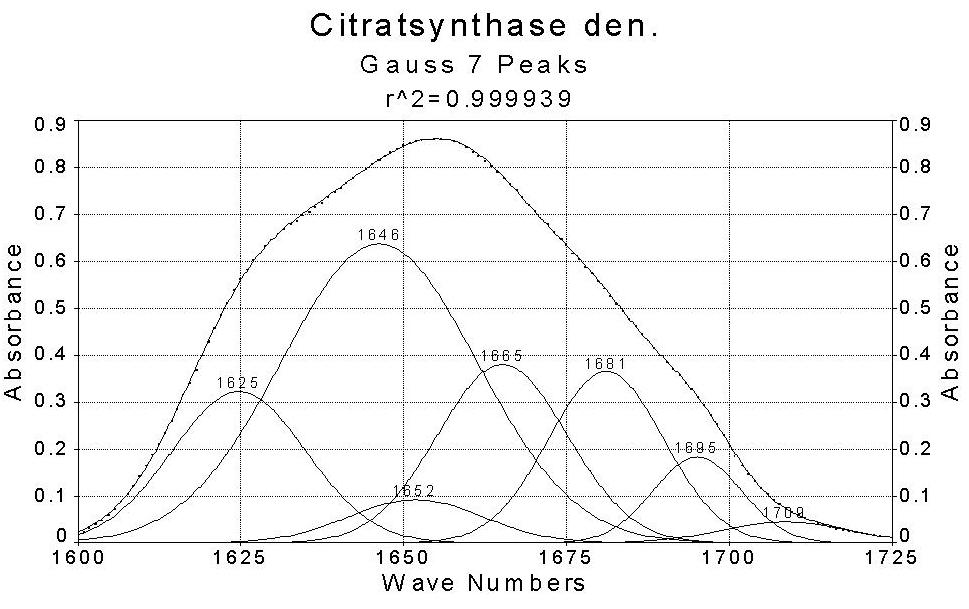 CSC
CSC
During the lastyears, the FTIR spectroscopy was used for the determination of secondary structures in
denatured proteins
and inclusion bodys
|
|
Beutenbergstraße 11 D-07745 Jena • Germany |
Phone: +49 3641 65-6000 Fax: +49 3641 65-6351 |
E-mail: info@leibniz-fli.de www.leibniz-fli.de |
Data Privacy Imprint |
 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}